Syndrome de Brugada - Définition
La liste des auteurs de cet article est disponible ici.
Introduction
| Syndrome de Brugada Classification et ressources externes | |
| |
|---|---|
| CIM-10 | I42.8 |
| CIM-9 | 746.89 |
| OMIM | 601144 |
| DiseasesDB | 31999 |
| eMedicine | med/3736 |
| MeSH | D053840 |
Le syndrome de Brugada est une maladie génétique rare caractérisée par un sus-décalage du segment ST au niveau des dérivations précordiales droites V1, V2 et V3, et un aspect de bloc de branche droit à l'électrocardiogramme associés à un risque élevé d'arythmie ventriculaire pouvant entrainer syncope et mort subite, sur un cœur structurellement sain. La transmission se fait sur un mode autosomique dominant et la pénétrance est variable. Des mutations génétiques entrainent des anomalies au niveau des canaux ioniques. L'âge moyen du premier épisode clinique est de 40 ans, avec une forte prédominance masculine. La prévalence estimée est d'environ 1/1000 dans les pays asiatiques, probablement plus faible ailleurs. Le pronostic est grave chez les patients présentant des symptômes et la mort subite peut être prévenue par la pose d'un défibrillateur automatique. Ce syndrome a été décrit pour la première fois en 1992 par les frères Pedro et Josep Brugada.
Historique
Le syndrome a été décrit pour la première fois par les frères Brugada.
Épidémiologie
La fréquence est estimée à 1 sur 1000 dans les populations asiatiques où le syndrome de mort brutale au cours du sommeil est fréquent.
L'âge moyen du diagnostic ou de la mort subite est de 40 ans plus ou moins 22 ans, avec des extrêmes allant entre deux jours de vie jusqu'à plus de 80 ans.
Il semble plus fréquent chez les hommes, ces derniers ayant des formes plus graves.
Étiologie
Le syndrome de Brugada est une canalopathie sodique. L'une des causes est une mutation du gène SCN5A 600163 (en) situé sur le chromosome 3. Près de 300 mutations différentes ont été identifiées sur ce gène mais ces mutations ne sont présentes que dans un cas de Brugada sur cinq. La mutation sur le gène de l'enzyme glycerol-3 phosphate dehydrogenase-1 like (GPD1-L) provoque une maladie semblable.
Il s'agit d'une maladie de transmission autosomique dominante avec une pénétrance faible (peu de porteurs de la mutation présentent les signes de la maladie).
La mutation du gène SCN5A n'est pas la seule cause de la maladie, en effet on peut être porteur d'un syndrome de Brugada (symptomatique ou non) sans avoir de mutation sur le gène SCN5A. Il est possible de mettre en évidence une mutation génétique du SCN5A sur seulement 10 à 25% des patients atteints d'un syndrome de Brugada.Pour les autres patients atteints sans avoir de mutation du SCN5A,la maladie est bien là, mais la mutation d'autres gènes non encore identifiés sont certainement en cause. Des recherches génétiques sont en cours et notamment par le Docteur Ramon Brugada à Montréal.
Diagnostic
Les éléments intervenant dans le diagnostic sont :
- Antécédents familiaux de Syndrome de Brugada, de mort subite ;
- Antécédents personnels de mort subite ressuscitée ;
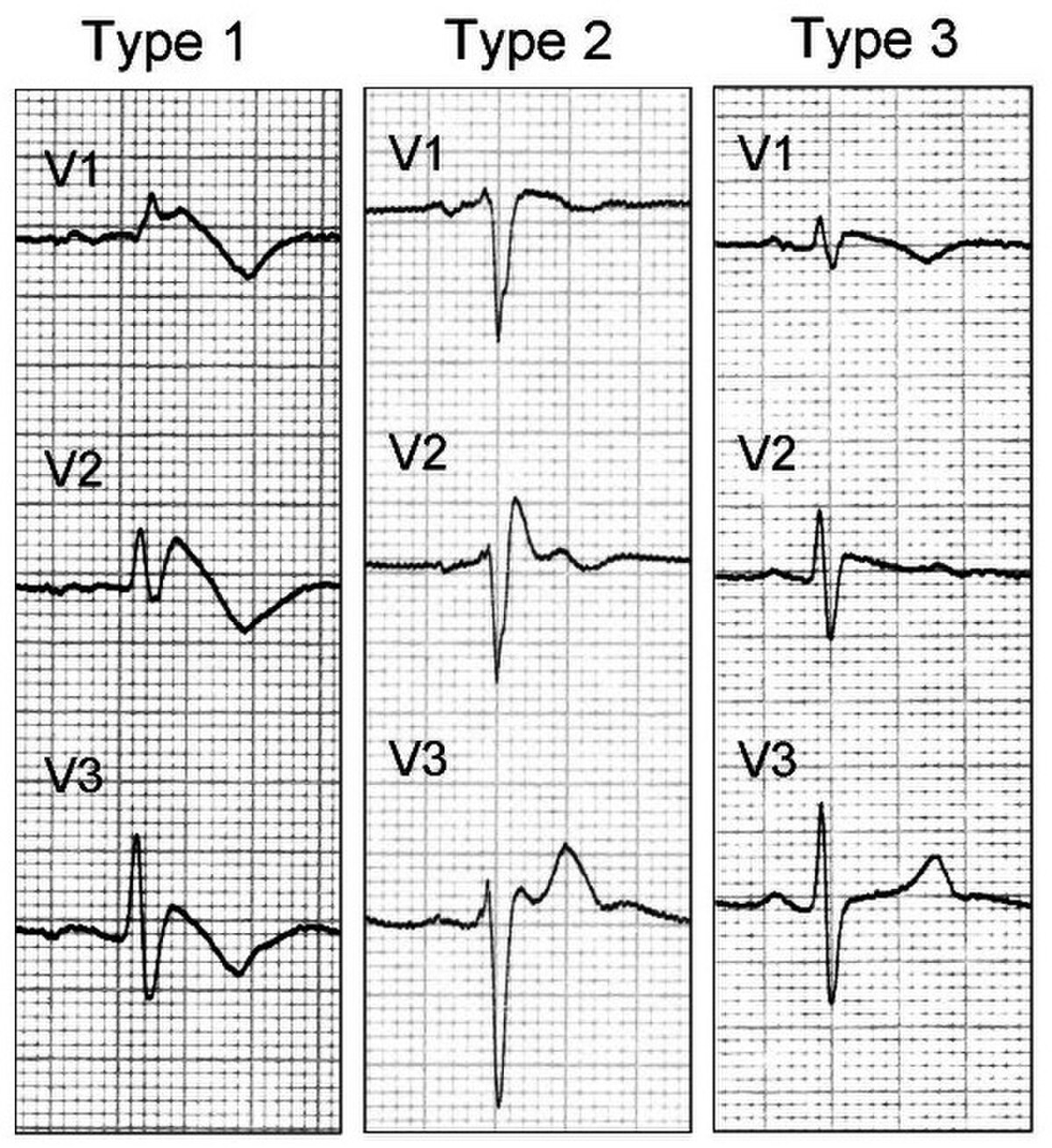
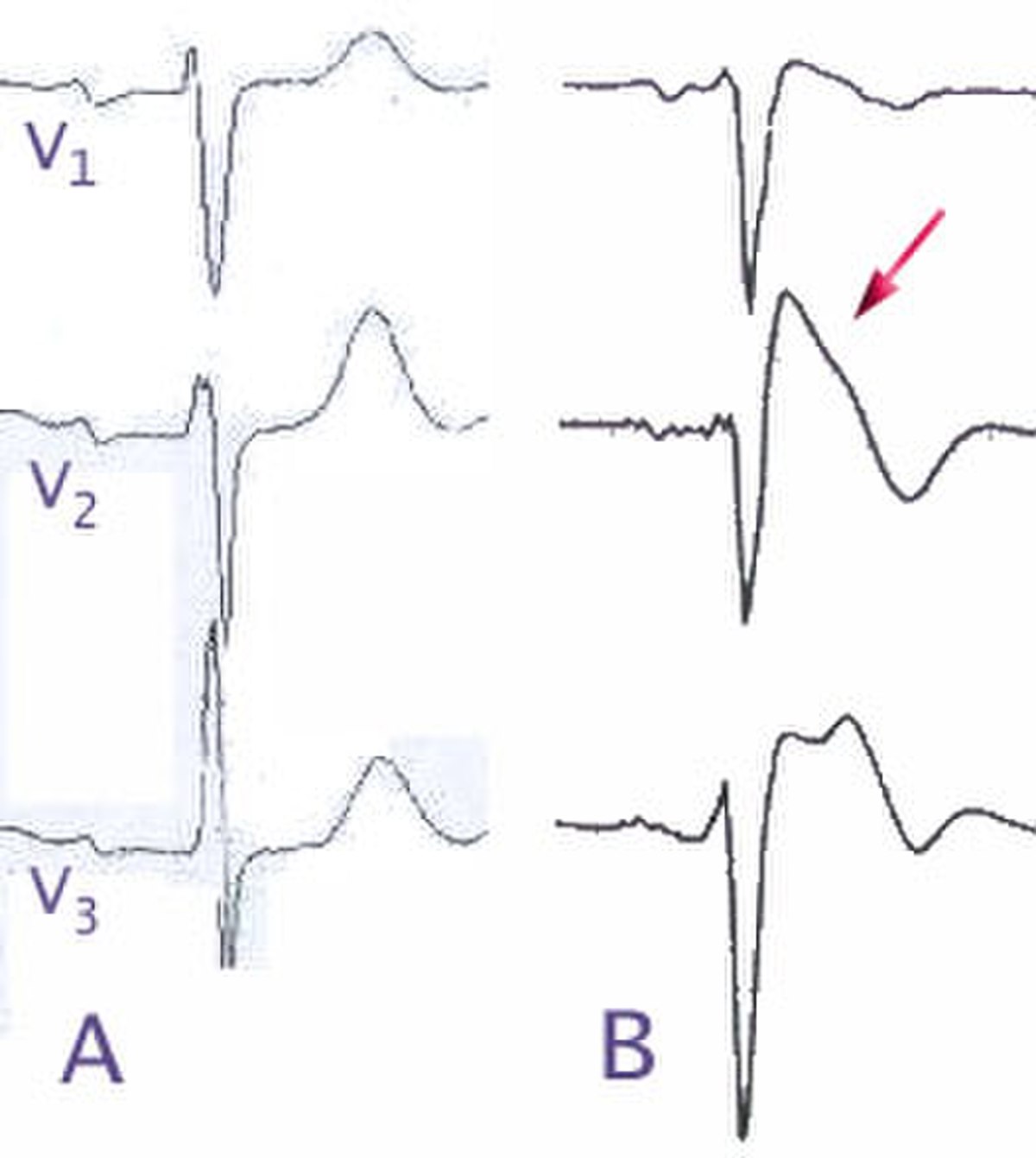
- A l'électrocardiogramme : aspect de bloc de branche droit associé à un sus-décalage du segment ST et des anomalies de l'onde T dans les dérivations précordiales droites (V1 à V3). On décrit 3 types d'anomalies ECG:
- Le type 1: sus-décalage du segment ST en forme de dome.
- Le type 2 : sus-décalage du segment ST en forme de selle de cheval.
- Le type 3 : même figure sur l'électrocardiogramme que le type 2 mais en aplatie.
- Positivité du test à l'Ajmaline ou à la Flécaïne ;
- Identification de la mutation du gène codant SCN5A.
- Électrocardiogramme