Séquençage des protéines - Définition
La liste des auteurs de cet article est disponible ici.
Détermination de la séquence
Le marquage chimique de l'extrémité N-terminal (NH2)
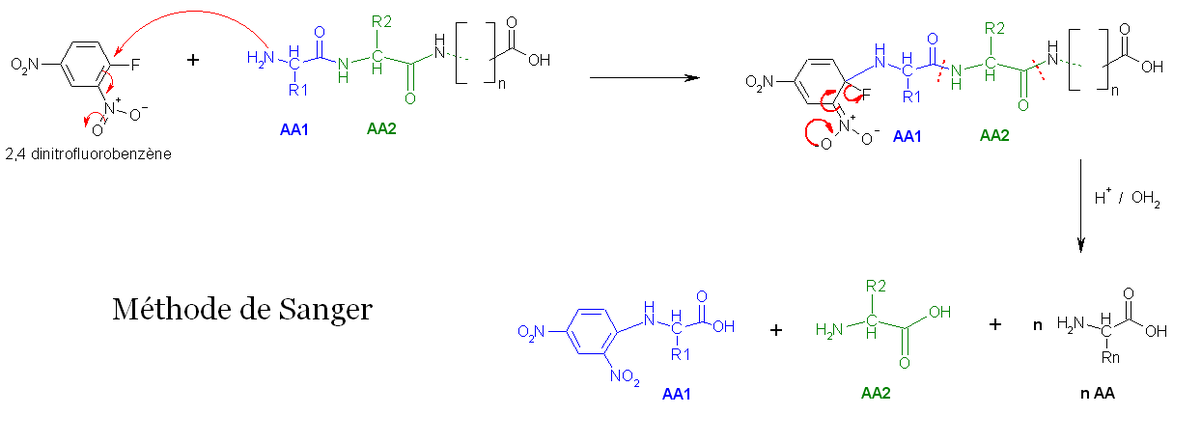
- La première méthode utilise la réaction de Sanger (cf. arylation). Les fonctions 2-amine des acides aminés et peptides réagissent avec le 2,4-dinitrofluorobenzène pour former des dérivés jaunes, les 2,4-dinitrophényl-peptides. Lorsque ces derniers ET (elle et constitue de different molecule)
composés sont soumis à une hydrolyse acide, toutes les liaisons peptidiques sont hydrolysées, mais la liaison entre la fonction 2,4-dinitrophényl et la fonction amine de l'acide aminé N-terminal restent stables, d'où l'identification de ce dernier.
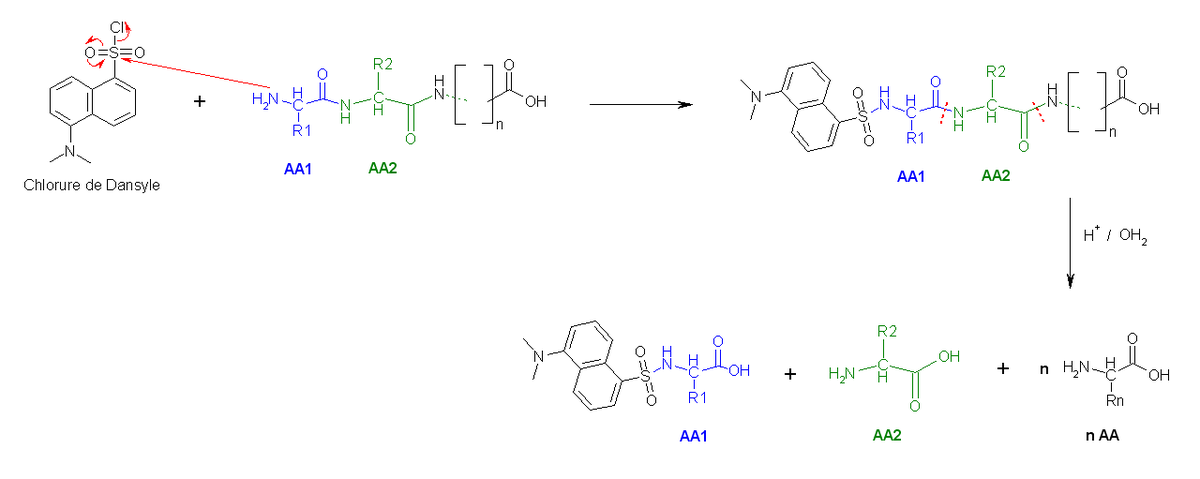
- La deuxième méthode utilise le chlorure de dansyle, qui, combiné à une hydrolyse totale acide forme un Dansyl-AcideAminé1 (DNS-aa) qui est fluorescent et un hydrolysat.
- Une troisième méthode est celle d'Edman, dans laquelle le phénylisothiocyanate (PITC) réagit avec la fonction amine N-terminale pour donner un complexe qui, après action d'un acide dans des conditions douces libère une phénylthiohydantoïne et le peptide restant intact (cf. carbamylation). Les deux premières méthodes utilisant une hydrolyse totale acide, le reste de la chaîne peptidique voit ses acides aminés clivés. Le grand avantage de la méthode d'Edman est que la chaîne peptidique reste intacte et peut être récupérée et soumise à nouveau au traitement avec le PITC.
Identification des acides aminés C-terminaux
On utilise l'hydrazynolise, qui coupe les liaisons à 100°C et donne un acide aminé libre. On peut également utiliser des carboxypeptidases qui coupent l'extrémité C-terminale, il en existe plusieurs types:
- la carboxypeptidase A, qui coupe tout sauf les acides aminés basiques (elle est bloquée par la proline)
- la carboxypeptidase B, qui est spécifique des acides aminés basiques (Lys, Arg)
Analyse enzymatique des extrémités N et C-terminaux
Il existe également des méthodes biochimiques utilisant des enzymes capable de couper le premier ou le dernier acide aminé de la chaîne polypeptidique, on les appelle des exopeptidases. Les aminopeptidases clivent la liaison peptidique située juste après le premier acide aminé et libèrent celui-ci. De manière symétrique, les carboxypeptidases clivent la liaison peptidique située juste avant le dernier acide aminé.
Il en existe plusieurs de ces enzymes de spécificités variées, par exemple parmi les carboxypeptidases, on trouve :
- La carboxypeptidase A, clive l'acide aminé C-terminal lorsque celui-ci est aromatique ou aliphatique
- La carboxypeptidase B, clive l'acide aminé C-terminal lorsque celui-ci est basique (Arginine ou Lysine)
- La carboxypeptidase Y à spectre large
En analysant les acides aminés libérés par ces enzymes, il est possible d'analyser la séquence N ou C-terminale de la protéine.
Fragmentation de la chaîne polypeptidique
La chaîne polypeptidique est découpée en une série de petits peptides par des hydrolyses enzymatiques ou chimiques.
Méthode chimique
L'hydrolyse chimique la plus utilisée implique une réaction avec le bromure de cyanogène, qui scinde les liaisons peptidiques dans lesquelles la fonction carboxyle est fournie par des résidus de méthionine.
Méthode enzymatique (utilisation d'endopeptidases)
L'hydrolyse enzymatique utilise les protéases, enzymes qui hydrolysent les liaisons peptidiques. La trypsine catalyse l'hydrolyse des liaisons peptidiques dans lesquelles la fonction carboxyle est fournie par la lysine ou l'arginine, la chymotrypsine permet la coupure du côté C-terminal des acides aminés aromatiques (Phe, Trp et Tyr).
- Trypsine (C) : , (sauf si à droite)
- Chymotrypsine (C) : , , (sauf si à droite)
- Thermolysine (N) : , , (sauf si à gauche)
- Pepsine (N) : , , (sauf si à gauche)
- LysC
- Papaïne : très utilisée dans l’industrie agro-alimentaire et chimique, elle permet notamment en boucherie d’attendrir la viande. Elle est aussi utilisée dans les produits de nettoyages de verres de contact, ainsi que dans les poudres à lessive, où son action est de détruire les protéines responsables des taches.
- Clostripaïne
- Subtilisine
- Elastase
- etc.
Identification de la séquence
L'identification de la séquence se fait de manière logique à partir des résultats obtenus par l'utilisation des différentes méthodes décrites précédemment.
Séquençage par spectrométrie de masse
La fragmentation par spectrométrie de masse MS/MS permet de séquencer des courtes séquences d'acides aminés (10 à 20). Couplée avec les techniques de digestion des protéines précédemment décrite, il est ainsi possible de connaitre la séquence d'une protéine. Cependant, cette approche nécessite d'utiliser différents protocoles de digestion différents pour espérer obtenir l'ensemble de la séquence de la protéine.
Pour un organisme dont le génome est entièrement connu, on utilisera un seul type de digestion (par exemple une digestion par la trypsine suivie d'une analyse par HPLC couplée à un spectromètre de masse de type ESI-MS/MS), ce qui permet d'obtenir jusque 30% de la séquence de la protéine (ce qui est suffisant pour caractériser la protéine).
Pour un organisme dont le génome n'est pas entièrement connu, on utilise en première intention un séquencage automatisé (séquençace d'Edman par exemple).

















































