Progéria - Définition
La liste des auteurs de cet article est disponible ici.
Introduction
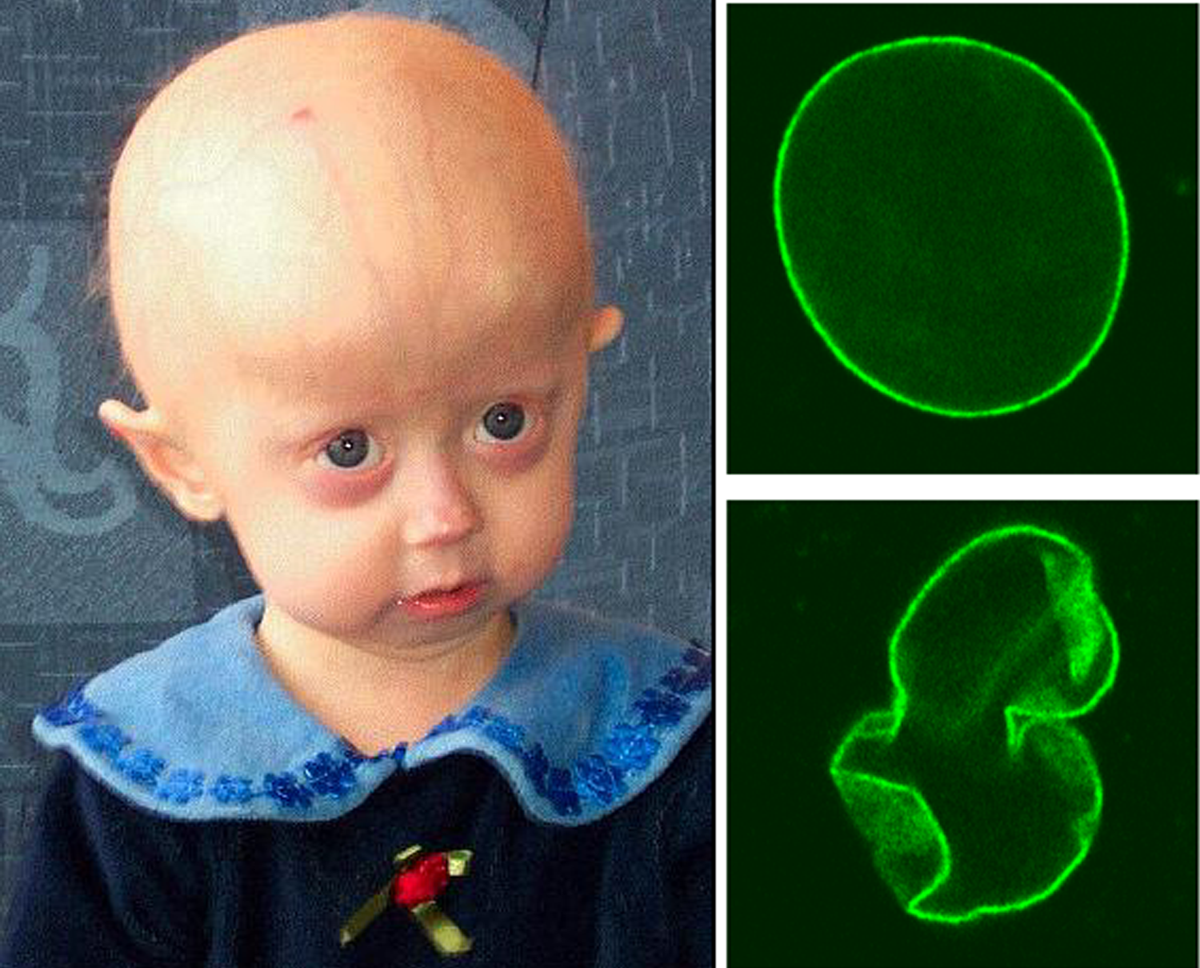
La progéria, ou Syndrome de Hutchinson-Gilford, est une maladie génétique extrêmement rare qui provoque des changements physiques qui ressemblent fort à une sénescence accélérée de ceux qui en sont atteints (vieillissement accéléré dès la première ou la deuxième année). Il n'y a pas de traitement spécifique connu.
Historique
Maladie rarissime décrite en 1886 par Jonathan Hutchinson et Hastings Gilford en 1904.
Causes biologiques
Depuis 2003, des scientifiques français et américains ont identifié le gène lamineA sur le chromosome 1 responsable du vieillissement prématuré des personnes atteintes. On observe chez les malades une mutation ponctuelle de substitution remplaçant une molécule de cytosine par une molécule de thymine en position 1824 dans le gène LMNA. La lamine A est une protéine structurale de la matrice nucléaire, qui sous forme de pré-lamine A se trouve dans l'enveloppe nucléaire, avant d'être clivée en lamine A qui diffuse dans le noyau cellulaire. Dans la forme mutée, la lamine A perd une cinquantaine d'acides aminés pour devenir une « progérine » qui ne peut migrer dans le noyau cellulaire : elle reste attachée à la membrane nucléaire, entraînant sa déformation et l'adhésion de la chromatine à cette dernière.
Le mécanisme précis entraînant le vieillissement prématuré reste cependant à élucider.
On peut par contre déjà dire que le mécanisme défectueux dans la progeria n'est pas une exacerbation d'un processus de vieillissement normal, mais un mécanisme qui semble complètement différent.
Il existe plusieurs souches de souris ayant une maladie proche de la progeria humaine, comportant soit une mutation sur le gène LMNA, soit une mutation sur le gène Zmpste24 empêchant le détachement de la lamine de la membrane nucléaire.
Symptômes
La maladie touche les deux sexes. L'apparition des symptômes débute entre 18 et 24 mois. Ils se manifestent par une croissance retardée, une alopécie, et une morphologie de la face caractéristique marquée par sa petitesse, de petites mâchoires, et un nez pincé. En évoluant, la maladie cause un vieillissement accéléré de la peau (rides, finesse), de l'athérosclérose, une ostéolyse des clavicules et des phalanges, des lipodystrophies, et des problèmes cardio-vasculaires. Des troubles musculaires et squelettiques apparaissent, la taille des malades ne dépassant pas 110 cm pour un poids de 15 kg. Le développement mental n'est en revanche pas affecté. La plupart des enfants atteints de cette maladie meurent entre l'âge de 13 et 16 ans d'un vieillissement prématuré (le record de longévité d'un malade est de 26 ans).
En revanche les patients ne présentent aucune maturation sexuelle prématurée, ni de cancers comme souvent pour le vieillissement naturel.
Traitement
La maladie est pour l'instant constamment mortelle avant l'âge adulte normal. Il existe cependant quelques pistes de recherche.
- La progérine resterait attachée à la membrane nucléaire en raison de la fixation d'un groupe farnésyl sur celle-ci. L'inhibition de cette « farnésylation », soit par diminution de la synthèse de ce composant, soit par diminution de la fixation de ce groupe, semble avoir une certaine efficacité sur la déformation du noyau.
- L'inhibition de la production en lamine pourrait être également une autre piste.
Actuellement (avril 2009), deux essais cliniques sont en cours pour tenter de ralentir la progression de la maladie chez les enfants atteints, l'un aux Etats-unis à Boston, l'autre en France, à Marseille (équipe du Pr Nicolas Lévy). Les américains testent une molécule utilisée en thérapie anti-cancer, qui bloque la fixation du groupe "farnésyle" sur la progérine. Cette molécule est de la famille des FTI (farnesyl-transferase inhibitor). http://www.clinicaltrials.gov/ct2/show/NCT00425607?term=progeria&rank=3
L'équipe marseillaise a démarré en octobre 2008 un essai prévoyant de traiter 15 patients pendant 2 ans, en combinant des injections régulières d'un amino-biphosphonate et la prise quotidienne d'un comprimé d'une statine. Les amino-biphosphonates sont utilisés principalement pour lutter contre l'ostéoporose de la femme ménopausée, mais aussi contre les atteintes osseuses dans certains cancers. Les statines sont le principal traitement prescrit contre l'hypercholestérolémie dans le monde. La combinaison de ces 2 médicaments vise à ralentir la fabrication du groupe farnésyle, donc à diminuer la quantité de progérine farnésylée produite. http://www.clinicaltrials.gov/ct2/show/NCT00731016?term=progeria&rank=4

















































