Prion (protéine) - Définition
La liste des auteurs de cet article est disponible ici.
Maladies
Troubles dus à sa présence
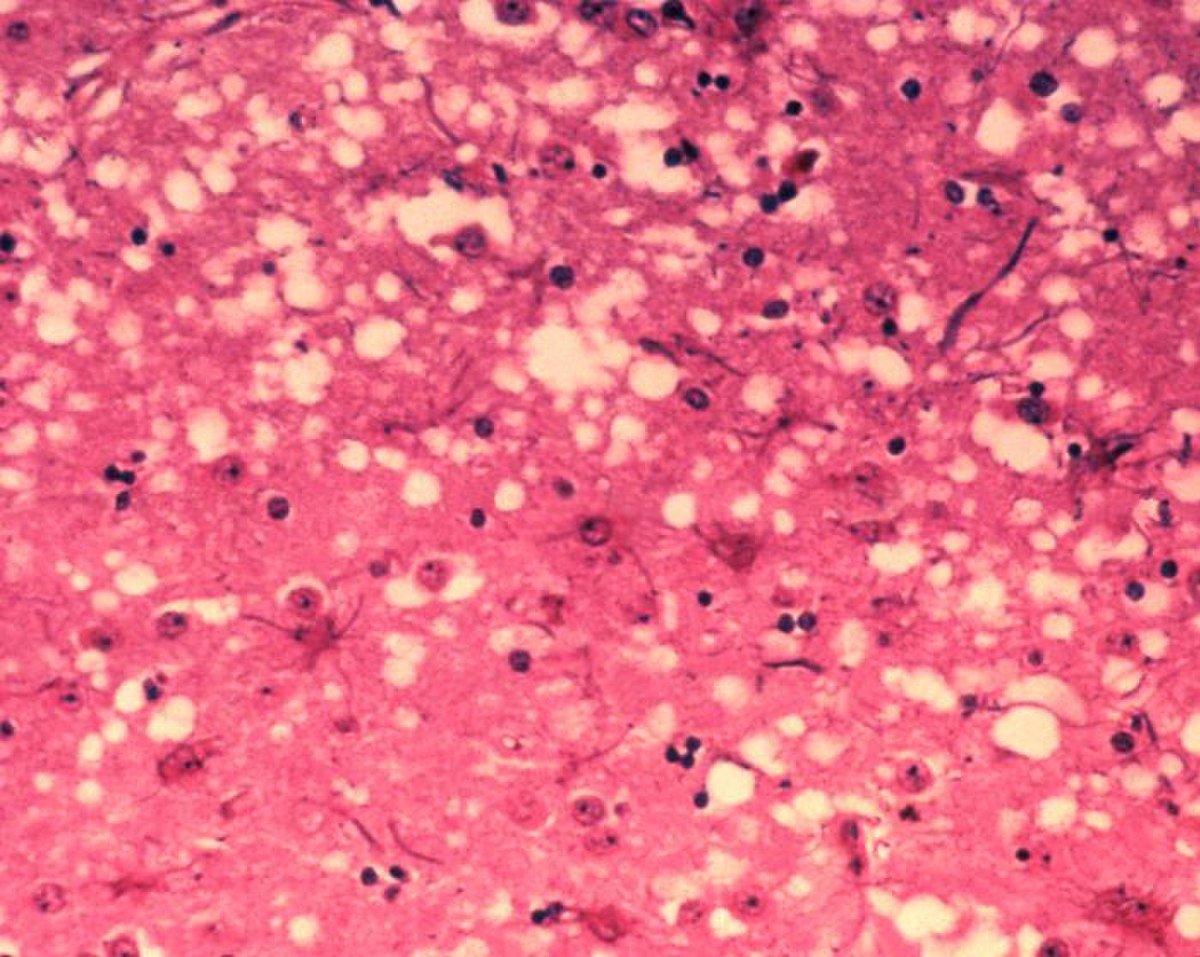
Les maladies à prions provoquent une dégénérescence du système nerveux central qui est toujours fatale.
- Le rôle de prions est établi dans certaines affections animales telles que l'encéphalopathie spongiforme bovine (ESB ou maladie de la vache folle), la tremblante du mouton et de la chèvre, et la maladie du dépérissement chronique des cervidés par Stanley Prusiner.
- Chez l’homme, il est responsable de la maladie de Creutzfeldt-Jakob qui se caractérise par une démence précoce aboutissant au décès. La forme commune est sporadique, atteignant le plus souvent le sujet âgé. Elle peut être rarement familiale, avec dans ce cas une implication du gène de la protéine prion. Elle peut être également transmise par inoculation de tissus contaminés (extraits d’hypophyse auparavant employés dans le traitement par l’hormone de croissance, greffes de cornée et de dure-mère, électrodes contaminées).
En mars 1996, est apparue une forme clinique chez le sujet jeune (< 30ans), appelé nouveau variant de la maladie de Creutzfeld-Jakob, dont le lien avec l’ESB a été prouvé ensuite. La transmission serait due probablement à l’ingestion de viande bovine contaminée par l’ESB. Le prion est également la cause d’autres maladies humaines : le kuru aujourd’hui disparu (touchant des tribus Foré de Nouvelle-Guinée qui avaient la particularité culturelle de manger le cerveau de morts lors de rites anthropophages mortuaires et qui a été la 1re encéphalopathie spongiforme humaine dont la transmissibilité au singe a été démontrée), la maladie de Gertsmann-Sträussler-Scheinker et l’insomnie fatale familiale.
Il existe d’autres maladies neurologiques comportant des accumulations de protéines anormales, telles la maladie d'Alzheimer et la maladie de Parkinson. La responsabilité d’un prion n’a toutefois pas été démontrée dans ces cas, bien qu’il puisse coexister.
Troubles dus à l’absence de Prp-c
Les données disponibles proviennent d’expérimentation de transgenèse sur des souris/hamsters à qui on a retiré le gène de la protéine Prp-c et qui donc ne possèdent plus cette protéine, ou dont on peut stopper à volonté la production de protéine Prp-c. Ces travaux permettent d’élucider peu à peu les fonctions de la protéine. Certaines souris dépourvues de protéine par knock-out du gène prnp codant cette protéine, sont viables et fertiles, sans phénotype apparent. D'autres développent une mort neuronale massive au niveau du cervelet. Cette mort est due à une autre protéine, paralogue à la protéine saine Prp-c, appelée Doppel (Dpl).
Ce sont plus des modèles expérimentaux que de véritables prions puisqu’il manque dans ces cas la notion d'«infection». Les « Prp-c » de levure ne forment pas des protéines prion comme chez les animaux, mais sont en réalité des protéines (souvent de choc thermique) qui en miment le comportement : dans certaines conditions de stress, elles changent de conformation et s'accumulent, perturbant le fonctionnement cellulaire de la levure.
Protéines normales et pathologiques
Le prion ou protéine Prp-sc est une forme spéciale de la protéine Prp-c qui est présente à l’état naturel et est impliquée dans le fonctionnement normal de la cellule. Les fonctions de Prp-c ne sont pas encore connues précisément mais on les soupçonne essentielles. En effet, la protéine Prp-c était présente avant la spéciation des mammifères, ce qui signifie que tous les mammifères (et donc l'homme) sont susceptibles de développer des maladies à prions. La protéine Prp-c est impliquée dans le développement du système nerveux chez l'embryon. Chez l'adulte, elle est exprimée essentiellement dans le cerveau et la moelle épinière (neurones et glie). Elle est impliquée dans les processus de différenciation et d’adhésion des cellules. Elle aurait aussi un rôle protecteur antioxydant et vis-à-vis de la mort cellulaire programmée (apoptose). Cette protéine aurait également un rôle dans le repliement d’autres protéines.
Selon l'équipe du Dr Scott (décembre 2006), la protéine normale, étudiée chez le rat, présente des accumulations particulières à l'intérieur des cellules du pancréas spécialisées dans la production d'insuline, et les rats prédisposés au diabète présentent 3 fois plus de cellules productrices d'insuline avec des amas de protéines Prp-c. Le taux de Prp-c dans le pancréas d'un rat normal change fortement dans les un à trois jours suivant l'administration de concentrations élevées de sucre via le sang. La protéine Prp-c pourrait être impliquée dans le diabète de type 1 ou juvénile, maladies caractérisées par une attaque par le système immunitaire des cellules produisant l'insuline (dans le pancréas).
Le prion est une protéine Prp-c repliée différemment, noté Prp-sc. La Prp-sc résulte d’une modification de la structure tridimensionnelle de Prp-c. Elle provoque les maladies à prions (maladie de la vache folle, ou encéphalopathie spongiforme bovine, maladie de Creutzfeldt-Jakob, tremblante du mouton, Chronical Wasting Disease ou maladie du dépérissement chronique des cervidés). Lors de l'infection, l'agent prion, agent pathogène responsable de l'infection, pénètre le neurone, où pour des raisons et par un mécanisme encore mal compris il se multiplie, en dépliant/repliant les protéines Prp-c en protéines Prp-sc, forme qui n'est plus dégradée par protéolyse et qui, par accumulation dans la cellule, finit par la tuer et former des plaques de dépots dans le cerveau.
Dans toutes ces maladies, aucun acide nucléique (ADN/ARN) n’a pu être spécifiquement associé à l’infectiosité, comme a pu l'être la protéine Prp-sc. On parle d'agent transmissible non conventionnel (ATNC).
Les maladies à prions sont transmissibles d’un individu à l’autre et dans une certaine mesure d’une espèce à l’autre.

















































