Principe Franck-Condon - Définition
La liste des auteurs de cet article est disponible ici.
Extension du principe Franck-Condon en spectroscopie
Le principe Franck-Condon, dans sa forme canonique, s'applique seulement aux modifications des niveaux vibrationnels d'une molécule dans le cas d'une modification des niveaux électroniques par absorption ou émission d'un photon. L'intuition physique qui sous-tend ce principe est guidée par l'idée que les coordonnées atomiques des atomes constituants la molécule n'ont pas le temps de changer durant la brève période de temps prise pour la transition électronique. Néanmoins, cette intuition physique peut être (et l'est) étendue de manière routinière aux interactions entre des molécules émettrices ou réceptrices de lumière (chromophores) et leur environnement. Les extensions du principe Franck-Condon sont appropriées dans la mesure où ces molécules interagissent parfois fortement avec les molécules les entourant, particulièrement dans les liquides et solides, et ces interactions modifient les coordonnées nucléaires du chromophore de manière tout à fait analogue aux vibrations moléculaires considérées dans le principe Franck-Condon.
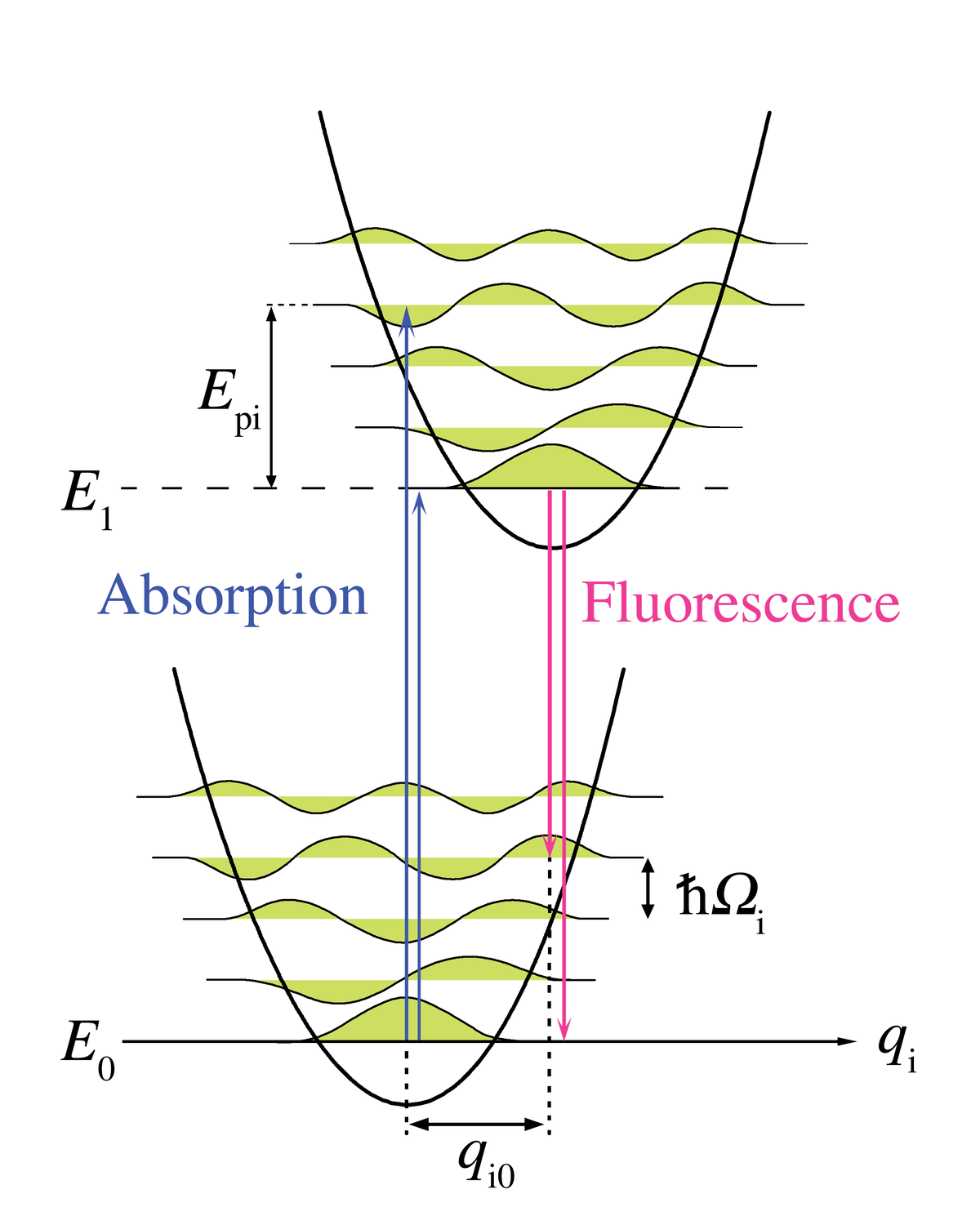
Principe Franck-Condon pour les phonons
L'analogie Franck-Condon la plus immédiate est due à l'interaction des phonons - quanta du réseau cristallin - avec les transitions électroniques des chromophores inclus dans le réseau comme impuretés. Dans ce cas, les transitions vers les niveaux électroniques les plus élevés peuvent se produire lorsque l'énergie du photon correspond à l'énergie d'une transition purement électronique ou à l'énergie d'une transition purement électronique plus l'énergie d'un ou plusieurs phonons de réseau. Dans l'approximation des températures basses, l'émission se produit du niveau zéro-phonon de l'état excité au niveau zéro-phonon de l'état fondamental ou vers des niveaux de phonons plus élevés de l'état fondamental. Comme pour le principe Franck-Condon, la probabilité des transitions impliquant des phonons est déterminée par la superposition des fonctions d'ondes phononiques dans les niveaux d'énergie initial et final. Pour le principe de Franck-Condon appliqué aux transitions de phonons, l'axe horizontal de la figure 1 est remplacé figure 5 par la coordonnée configurationnelle pour un mode normal. L'énergie potentiel du mode de réseau qi dans la figure 5 est représentée comme celle d'un oscillateur harmonique, et l'espacement entre niveaux de phonons (

Principe Franck-Condon pour la solvatation
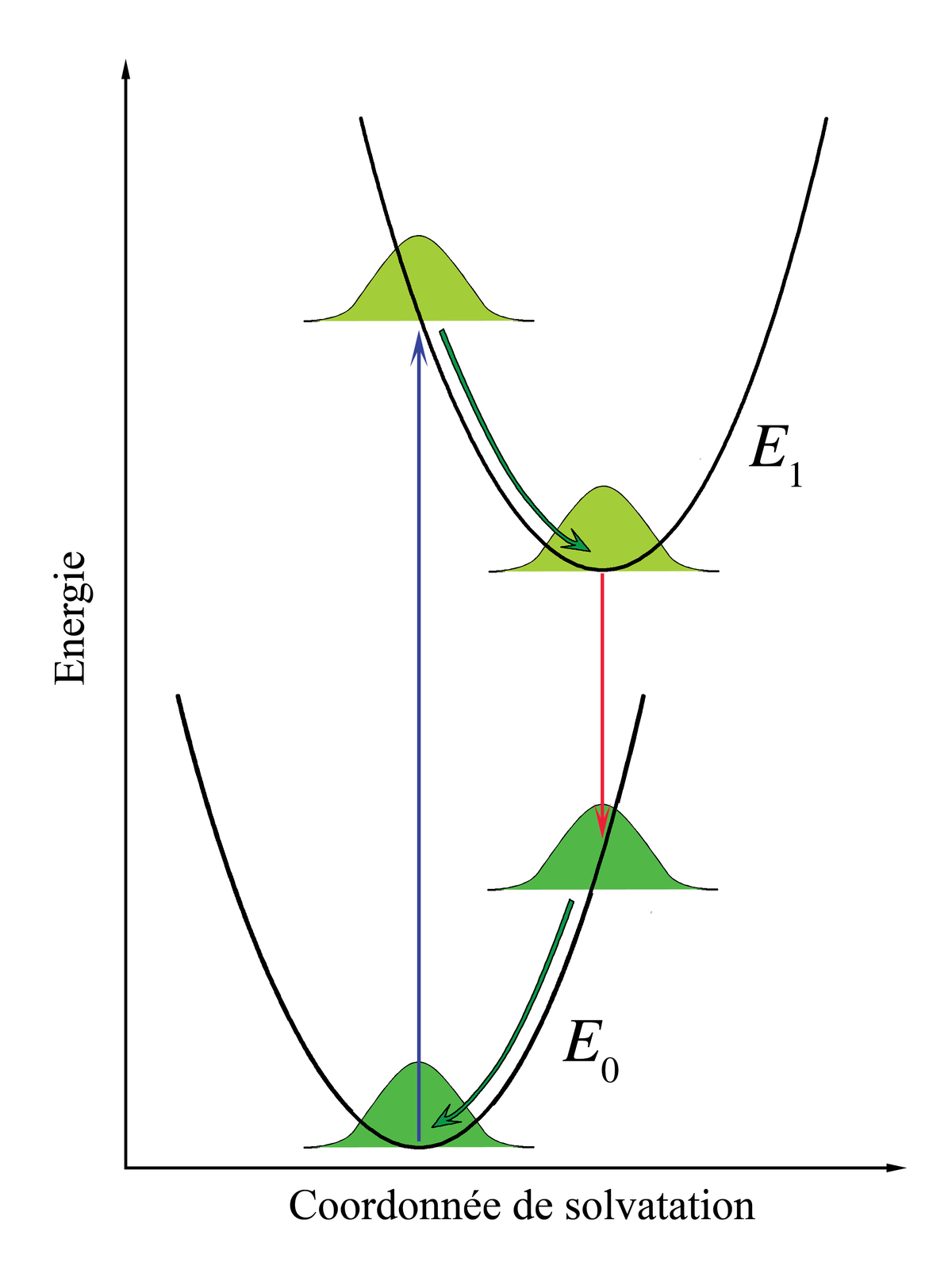
Les considérations Franck-Condon peuvent être aussi appliquées aux transitions électroniques des chromophores dissouts dans des liquides. Dans cette perspective, les niveaux vibrationnels des chromophores, comme les interactions des chromophores avec les phonons du liquide, continuent à la structure des spectres d'absorption et d'émission, mais ces effets sont considérés comme séparés et indépendants.
Considérons les chromophores comme entourés par les molécules du solvant. Ces molécules peuvent interagir avec les chromophores, particulièrement si les molécules du solvant sont polaires. L'association entre le solvant et le soluté est appelée solvatation et est une interaction stabilisante, ce qui signifie que les molécules de solvant peuvent se déplacer et tourner jusqu'à ce que l'énergie d'interaction soit minimisée. L'interaction en elle-même mets en jeu des forces électrostatiques et de van der Waals et peut parfois comprendre des liaisons hydrogène. Les principes Franck-Condon peuvent être appliqués lorsque les interactions entre le chromophore et les molécules de solvant l'entourant sont différentes entre états fondamentaux et excités. Cette modification dans l'interaction peut se produire, par exemple, en raison d'une différences des moments dipolaires entre les deux états. Si le chromophore initialement dans son état fondamental est proche de l'équilibre avec les molécules le solvatant puis absorbe un photon qui l'excite, son interaction avec le solvant sera éloignée de l'équilibre dans l'état excité. Cet effet est analogue avec le principe Franck-Condon original : la transition électronique est très rapide comparée avec le mouvement des noyaux - le réarrangement des molécules de solvant dans le cadre d'une solvatation. On peut alors parler de transition verticale, mais la coordonnée horizontale est maintenant l'espace d'interaction solvant-soluté. Cet axe de coordonnée est parfois appelé « axe de solvatation » et représente, plus ou moins abstraitement, tout ce qui correspond aux dimensions pertinentes du mouvement de l'ensemble des molécules de solvant interagissant.
Dans le principe Franck-Condon original, après la transition électronique, les molécules se trouvant dans des états vibrationnels plus énergétiques commencent immédiatement à relaxer vers l'état vibrationnel le plus bas. Dans la cas de la solvatation, les molécules de solvant essaieront immédiatement de se réarranger afin de minimiser l'énergie d'interaction. Le taux de relaxation du solvant dépend de sa viscosité. Considérant que le temps de relaxation du solvant est court comparé au temps de vie de l'état électronique excité, l'émission se fera depuis l'état d'énergie le plus bas de l'état électronique excité du solvant. Pour des solvants à petites molécules, comme l'eau ou le méthanol à température ambiante, le temps de relaxation du solvant est de l'ordre de quelques dizaines de picosecondes alors que les temps de vie des états excités des chromophores vont des quelques picosecondes à quelques nanosecondes. Immédiatement après la transition vers l'état électronique fondamental, les molécules de solvant doivent aussi se réarranger afin de s'accorder à la nouvelle configuration électronique du chromophore. La figure 6 illustre le principe Franck-Condon appliqué à la solvatation. Lorsque la solution est éclairée par une lumière correspondant à l'énergie de transition électronique, certains chromophores se trouveront dans l'état excité. Dans ce groupe, il y aura une distribution statistique des énergies d'interaction solvant-chromophore, représentée dans la figure par une fonction de distribution gaussienne. L'interaction solvant-chromophore est dessinée comme étant un potentiel parabolique dans les deux états électroniques. Les transitions électroniques étant essentiellement instantanées sur l'échelle de temps du mouvement du solvant (flèche verticale), l'ensemble des chromophores dans un état excité est immédiatement loin de l'équilibre. Le réarrangement des molécules de solvant selon la nouvelle courbe d'énergie potentielle est représentée par le flèches courbes dans la figure 6. Remarquons que lorsque les transitions électroniques sont quantifiées, l'énergie d'interaction chromophore-solvant est traitée comme un continuum classique en raison du nombre important de molécules concernées. Bien que l'émission soit décrite comme allant se produire à partir du minimum du potentiel d'interaction solvant-chromophore de l'état excité, une émission significative peut se produire avant d'atteindre l'équilibre lorsque la viscosité du solvant est importante ou que le temps de vie de l'état excité est court. La différence d'énergie entre les photons émis et absorbés décrits dans la figure 6 est la contribution de la solvatation au déplacement de Stokes.

















































