Neurofibromatose de type I - Définition
La liste des auteurs de cet article est disponible ici.
Introduction
Cet article porte sur la neurofibromatose de type I qui n'a aucun rapport clinique et génétique avec la neurofibromatose de type II
| ||||
|---|---|---|---|---|
| Autre nom | Maladie de Recklinghausen | |||
| Référence MIM | 162200 | |||
| Transmission | Dominante | |||
| Chromosome | 17q11.2 | |||
| Gène | NF1 | |||
| Empreinte parentale | Non | |||
| Mutation | - | |||
| Mutation de novo | 50 % | |||
| Nombre d'allèles pathologiques | Plus de 500 connues | |||
| Anticipation | Non | |||
| Porteur sain | sans objet | |||
| Incidence | 1/4000 | |||
| Prévalence | 1/5000 | |||
| Pénétrance | 100 % | |||
| Nombre de cas | Inconnu | |||
| Maladie génétiquement liée | Syndrome de Watson | |||
| Diagnostic prénatal | Possible | |||
| Article principal | Phacomatose | |||
| Liste des maladies génétiques à gène identifié | ||||
La neurofibromatose de type I est aussi appelée maladie de Recklinghausen du nom du médecin allemand, Friedrich Daniel von Recklinghausen, qui le premier a décrit cette maladie en 1881. Elle est une des plus fréquentes maladie génétique à transmission autosomique dominante. Le gène NF1 est un des gènes dont le taux de mutation spontanée est un des plus importants chez l'homme : environ la moitié des personnes atteintes par cette maladie est le résultat d'une mutation de novo.
Épidémiologie
Son incidence est estimée à 1/4 000 et sa prévalence à 1/5 000.
Symptômes
Des taches "café au lait"
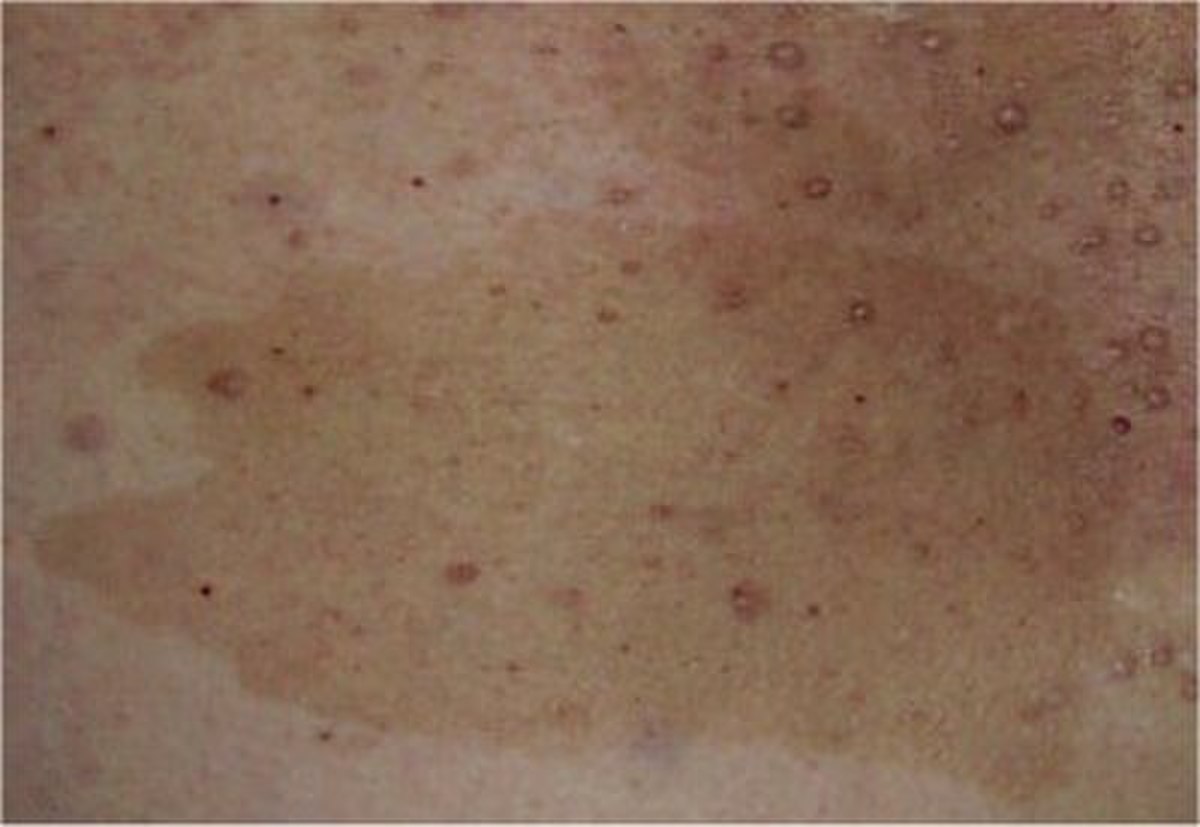
Ce sont des taches de couleur variant du beige clair au marron foncé et dont la taille varie pour un adulte de quelques millimètres à plusieurs centimètres.
Ces taches sont très fréquentes dans la population saine (plus de 10% de la population en possède une ou deux), mais dans le cas de la NF-1 leur nombre est de 6 au minimum, certains malades en ayant parfois plusieurs dizaines. Ces taches peuvent disparaître à l'âge adulte.
Elles apparaissent souvent dès la naissance. Pour être comptabilisées, elle doivent mesurer au minimum 5 mm chez un enfant et 1,5 cm chez un adulte.
Des neurofibromes
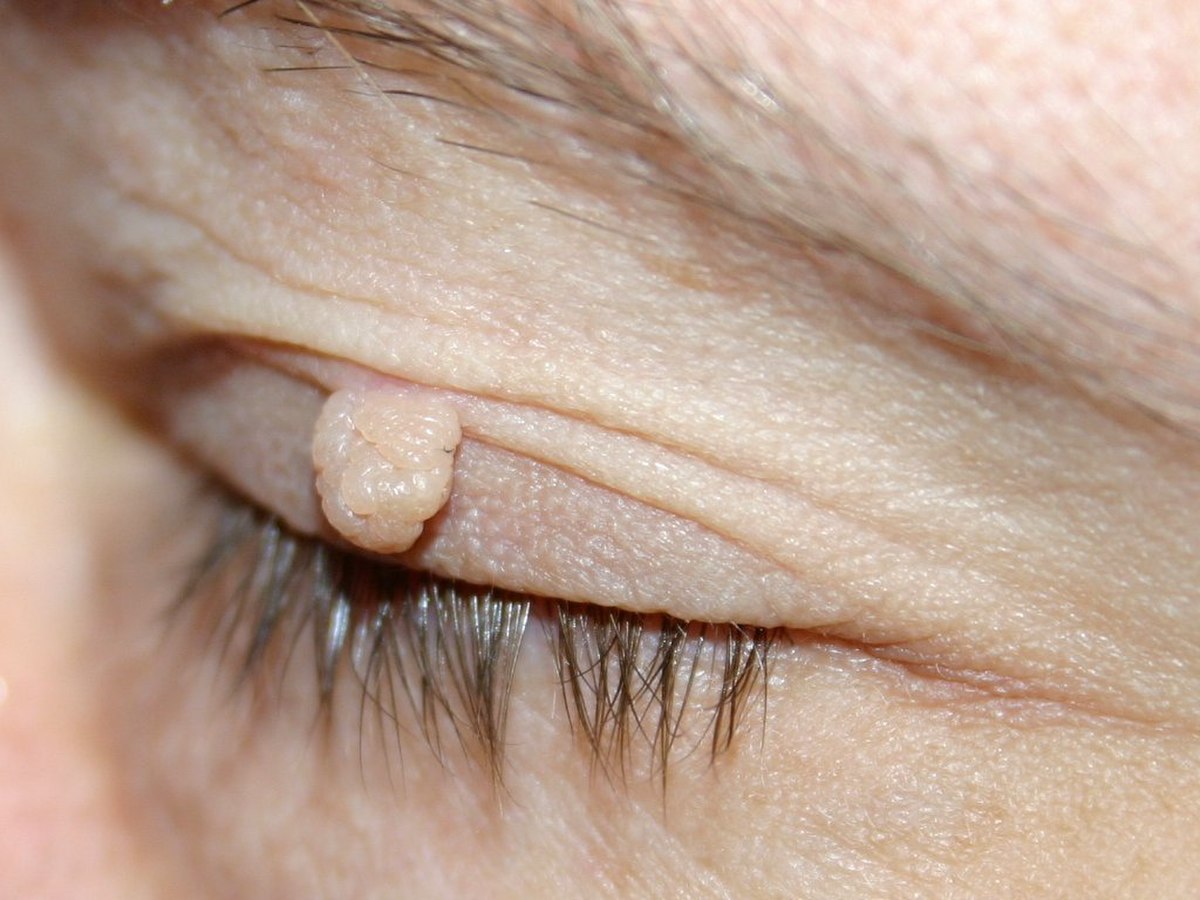
Ce sont des tumeurs bénignes issues des filets nerveux. On distingue trois type de neurofibromes :
- Les neurofibromes cutanés : ils ont un aspect de pseudo-hernie, ils sont de la couleur de la peau ou un peu plus rosés, sont dépressibles au toucher, et leur palpation n'entraîne en général aucune douleur. Leur taille est très variable d'un individu à l'autre, et ils n'apparaissent qu'à partir de la puberté. Ils peuvent être enlevés soit par la chirurgie classique soit au laser.
- Les neurofibromes nodulaires : petites tumeurs sous-cutanées qui poussent le long du trajet d'un nerf, en chaînette, ils ont l'aspect de ganglions, et sont douloureux au toucher.
- Les neurofibromes plexiformes : Ce sont des tumeurs souvent de grande taille, qui peuvent être invasifs et donc poser des problèmes de compression de certains organes vitaux. Ils apparaissent très tôt dans la vie, mais leur croissance au moment de la puberté les fait remarquer. Ils sont difficilement opérables car très vascularisés, et situés parfois à l'intérieur du corps.
Des lentigines
Ce sont de petites taches comme des taches de rousseur, mais leur localisation est caractéristique : creux de l'aisselle ou creux de l'aine. Elles sont présentes chez 80% des personnes atteintes de NF-1.
Des nodules de Lisch
Ce sont de petits nodules colorés situés dans l'iris. Difficiles à voir à l'œil nu, on les observe à l'aide d'une lampe à fente. Ils n'entraînent aucun trouble de la vision de l'œil. Ils sont en revanche un bon élément de diagnostic puisque 90% des adultes atteints de NF-1 en ont. Histologiquement, le nodule de Lisch est un hamartome.
Déformations osseuses caractéristiques
Il existe une courbure importante et un amincissement de la corticale des os longs, le plus souvent tibia, mais aussi fémur, radius, cubitus, ainsi qu'une résorption de l'os alvéolaire pouvant entrainer la chute des dents .

















































