Mucoviscidose - Définition
La liste des auteurs de cet article est disponible ici.
Diagnostic et dépistage
Diagnostic
- Critères diagnostiques
La suspicion de mucoviscidose se base sur les signes cliniques suivants mais cette liste n'est pas limitée :
- Des maladies pulmonaires ou bronchiques chroniques, récidivantes (toux chronique, encombrement et syndrome obstructif chronique, colonisation par certains germes, anomalies radiologiques, sinusites chroniques, hippocratisme digital)
- Des troubles digestifs (iléus méconial, prolapsus rectal, insuffisance pancréatique et syndrome de malabsorption, maladies hépatobiliaires chroniques)
- Une azoospermie,
- Un syndrome de perte de sel…
Le diagnostic est établi chez une personne présentant un ou plus des signes cliniques précédents associés à au moins un des signes de mauvais fonctionnement du gène CFTR suivants :
- Présence de mutation anormale du gène CFTR sur chacun des chromosomes 7, ou
- Deux tests de la sueur positifs (>60 mEq/L), ou
- Une recherche de différence de potentiel de l'épithélium nasal positive.
En l'absence de signe clinique, le diagnostic peut être fait :
- si un enfant est porteur de la même mutation du gène CFTR sur chacun des chromosomes 7 qu'un de ses frères et sœurs malades de la mucoviscidose,
- au cours d'un dépistage néo natal systématique (en 2002, 12,8 % des mucoviscidoses sont diagnostiquées de cette façon aux États-Unis),
- au cours d'un dépistage in utero (en 2002, 4 % des mucoviscidoses sont diagnostiquées pendant la période prénatale aux États-Unis).
Test de la sueur
Le test de la sueur est le dosage du chlore contenu dans la sueur du patient et représente un des principaux test diagnostique de la mucoviscidose. Il comprend trois temps : l'obtention de la sueur, le recueil de la sueur et la détermination de la concentration en chlorure.
La méthode de référence pour obtenir de la sueur est la stimulation des glandes sudoripares, au niveau de la face interne du bras, au-dessus du pli du coude, par iontophorèse à la pilocarpine: le passage d’un faible courant à travers la peau entre deux électrodes permet de faire passer la pilocarpine - molécule possédant des propriétés cholinergiques - à travers la peau, stimulant ainsi la sécrétion de sueur par les glandes sudoripares.
La méthode de référence pour le recueil de la sueur, délicat, est le recueil sur papier filtre, décrite par Gibson et Cooke. Il faut recueillir un échantillon d'au moins 100 mg de sueur. L'âge minimum requis est de 5 semaines avec un poids corporel d'environ 4 kg. Cet examen indolore dure 15 à 30 minutes.
Les méthodes utilisées pour déterminer la concentration en chlorures de la sueur sont de trois catégories : les méthodes quantitatives qui mesurent avec précision le poids de la sueur recueillie, les méthodes semi-quantitatives qui déterminent la concentration en électrolytes avec une approximation du volume recueilli et les méthodes qualitatives basées sur des réactions chimiques de précipitation permettent une estimation qualitative de la concentration en chlorures.
Chez les malades, les glandes sudoripares ont une teneur augmentée en ions Na+, K+ et Cl-. On ne teste cependant que la teneur en chlorure, plus discriminative que la teneur en sodium pour poser le diagnostic.
Un test de la sueur retrouvant une concentration de chlore supérieure a 60 mmol/l (millimoles par litre) est dit positif et est en faveur du diagnostic de mucoviscidose, il doit être positif à deux reprises pour affirmer le diagnostic. Un test retrouvant une concentration de chlore intermédiaire comprise entre 40 mmol/l et 60 mmol/l est douteux et suggère le diagnostic sans pour autant le poser avec certitude, des examens complémentaires sont nécessaires. Une concentration inférieure à 40 mmol/l est normale et le risque de mucoviscidose est faible.
Ce test est sensible, positif plus de 90% des cas de mucoviscidose. Il n'y a pas de corrélation entre la valeur retrouvée lors du test et la gravité de l'atteinte. Le test ne permet pas d'identifier les sujets hétérozygotes qui transmettent la maladie même si ceux-ci ont statistiquement des concentrations intermédiaires.
Les résultats supérieurs à 160 mmol/l ne sont pas physiologiquement possibles et sont à rapporter avec un problème technique.
Les faux positifs - le test est positif mais la personne n'est pas malade - exceptionnels, peuvent se retrouver dans le syndrome de Hurler, la fucosidose, la glycogénose de type 1, l'insuffisance surrénalienne aiguë, un déficit en alpha 1-antitrypsine, une hypothyroïdie, une dysplasie ectodermique, un diabète insipide, une néphronophtise, une anorexie, une dysfonction du système nerveux autonome, une maladie coeliaque, une cholestase familiale, une hypogammaglobulinémie, une hypoparathyroïdie, un syndrome de Klinefelter, une malnutrition, une mucopolysaccharidose de type 1, un pseudohypoaldosternonisme.
À l'inverse il existe de rares cas de mucoviscidose avec un test de la sueur douteux ou négatif. Quelques mutations sont associées à des résultats limites telles que R117H, 3849 + 10kb C-T, R334W ou P67L. Très rarement, le test peut même être normal chez un patient présentant un génotype mucoviscidosique avéré. De plus environ 1 à 2 % des malades ont une concentration de chlore inférieure à 60 mmol/l. Dans cette situation, seule l'identification de deux mutations sur le gène CFTR permet un diagnostic de certitude.
Différence de potentiel de l'épithélium nasal
Cet examen proposé par Knowles puis standardisé consiste à mesurer la différence de potentiel (DDP) de l'épithélium nasal et permet l'exploration in vivo des transports ioniques transépithéliaux. Il peut être utilisé à partir de 6 ans et il est réalisé dans de nombreux centres dans le monde dans le cadre de la démarche diagnostique. La mesure de la DDP nasal est accompagnée par des tests pharmacologiques associés destinées à mieux différencier la population malade de la non malade.
La DDP nasal n'est pas corrélée avec le test de la sueur ou avec la sévérité de la maladie ; et cette méthode ne détecte pas les sujets hétérozygotes. La valeur basale de la DDP nasal est modifiée par des pathologies telles l'inflammation, l'infection ou la polypose nasale.
Les individus atteints de mucoviscidose comparés à des individus indemnes ont :
- Une différence de potentiel élevée (plus négative) reflétant une absorption accrue du sodium,
- Une variation plus importante de la différence de potentiel durant l'administration au niveau de la muqueuse nasale d'amiloride, un inhibiteur de l'absorption sodique,
- Des variations minimes de la différence de potentiel sous perfusion de d'amiloride, de solution sans chlore et d'un agoniste ß2.
La mesure de la DDP nasal avec stimulations pharmacologiques dans le bilan de la mucoviscidose n'a pas d'intérêt dans les formes typiques de la maladie mais elle est intéressante dans les formes cliniques atypiques associées à des tests de la sueur « limites ».
Génotypage
Le génotypage par analyse génétique à partir d'un échantillon de sang vise à rechercher la présence d'une mutation anormale sur le gène CFTR. Il est essentiellement utilisé lors de la démarche diagnostique lorsque le test à la sueur n'est pas réalisable ou non informatif. Il peut être utilisé en première intention dans les cas suivants:
- Dépistage prénatal chez un fœtus à haut risque,
- Dépistage prénatal chez un fœtus à faible risque mais avec une hyperéchogénicité digestive fœtale,
- Dépistage néonatal,
- Enfants symptomatiques (avec un iléus méconial) trop jeunes (moins de 5 semaines) pour pouvoir réaliser un test de la sueur (impossibilité de collecter une quantité suffisante de sueur),
- Dépistage de la fratrie d'un patient atteint chez qui on a identifié les deux mutations du gène CFTR.
L’analyse génétique par la méthode de détection ciblée recherche les mutations parmi les plus fréquentes. En 2004, l'American College of Medical Genetics (ACMG) recommandait des kits analysant la présence de 23 mutations parmi les plus fréquentes, permettant d'atteindre un taux de détection allant de 57% à 97% selon les populations étudiées. D'autres méthodes telles la recherche de réarrangements, délétions, insertions, duplications ou la méthodes de balayage d’exons permettent de compléter l'analyse.
Chez un homme avec azoospermie ou oligospermie
La recherche d'une hypoplasie voire une agénésie des vésicules séminales et/ou des déférents par un urologue ou par d'imagerie médicale est la première étape. Si celle-ci est positive la recherche de mutation du gène CFTR est réalisée.
Diagnostic différentiel
Face à des éléments cliniques évoquant la mucoviscidose, se pose le problème d'un diagnostic différentiel avec d'autres maladies telles que :
- Une dilatation des bronches due à d'autres étiologies telles qu'un déficit immunitaire, un syndrome d'immobilité ciliaire, des séquelles de virose pulmonaire sévère (adénovirus), une pneumopathie négligée (tuberculose), une dysplasie broncho-pulmonaire ;
- Un wheezing chronique, un asthme ;
- Un tabagisme passif pouvant entraîner des manifestations respiratoires peu spécifiques ;
- Une maladie de Hirschsprung ou un syndrome du bouchon méconial, dans les formes de mucoviscidose avec occlusion intestinale néonatale associée ;
- Un déficit en alpha-1-antitrypsine, avec emphysème pulmonaire et signes respiratoires ;
- Un syndrome de dyskinésie ciliaire dont la maladie de Kartagener, exceptionnelle ;
- Un syndrome de Shwachman-Diamond ;
- Des pancréatites aiguës récidivantes ;
- Une insuffisance pancréatique chronique.
Dépistage
Dépistage néo-natal systématique
Les arguments en faveur d'un dépistage systématique sont:
- L'existence d'un test fiable,
- Le diagnostic parfois tardif de la maladie en raison des formes frustres,
- Le bénéfice d'un diagnostic et d'une prise en charge précoce des enfants atteints : plus la prise en charge - nutritionnelle essentiellement - est précoce, meilleur est le pronostic.
Il se déroule en deux étapes :
- Le test consiste à doser la trypsine immuno–réactive (TIR), proenzyme sécrétée par le pancréas circulant dans le sang. Il est réalisé trois jours après la naissance de l'enfant à partir d'une goutte de sang séchée en même temps que le dépistage de la phénylcétonurie, de l'hypothyroïdie et de l'hyperplasie congénitale des surrénales.
- La détection d'un niveau anormalement élevé de trypsine (>60 μg/l) conduit à réaliser un diagnostic par génotypage à la recherche des mutations CFTR les plus fréquentes. Le test génétique nécessite au préalable l'accord écrit et signé des parents (en France la loi traitant de l’analyse de l’ADN à des fins médicales est le Décret n° 2000-570 du 23 Juin 2000).
Les enfants chez lesquels une anomalie génétique (une ou deux mutation CFTR) est mise en évidence subiront dans le mois suivant leur naissance un test à la sueur pour déterminer leur statut de malade ou de porteur hétérozygote. Si aucune mutation n’est retrouvée, un contrôle de la trypsine est fait à J21.
Tous les programmes de dépistage utilisent maintenant ce test couplé TIR-ADN. La recherche des mutations comprend au minimum la mutation ΔF508 mais utilise le plus souvent des kits avec 20, 30 voire 50 mutations, représentant plus de 90 % des mutations existantes. Ces programmes de dépistage ne sont pas encore figés et sont évolutifs afin de devenir le plus efficace possible.
- En France
La France a été le premier pays à instaurer un programme de dépistage néonatal systématique fin 2002, dans les 3 premiers jours de vie à la maternité. Entre fin 2002 et fin 2005, 2 717 992 nouveau-nés ont bénéficié de ce programme. Une TIR élevée a été retrouvée chez 18 610 nouveau-nés (soit 0,7 % du total) ; 18 054 (soit 98 % d’entre-eux) ont bénéficié d'une recherche des principales mutations du gène CFTR ; 3 469 enfants (soit 1,3 pour 1000 nouveau-nés testés) ont été adressés à un CRCM (Centre de Références et de Compétences de la Mucoviscidose) parce qu’ils avaient 1 ou 2 mutations (n=2003) ou parce qu’ils avaient une hypertrypsinémie persistante à J21 (n=1466) ; 621 mucoviscidoses ont ainsi été diagnostiquées soit une incidence de 1 pour 4376 naissances, très variables selon les régions (de 1/2749 en Bretagne pour la plus forte à 1/7077 en Midi-Pyrénées pour la plus faible). Le kit CF30 détectait 87 % des mutations.
Le 26 avril 2007, le Comité consultatif national d'éthique (CCNE) français a émis un avis s'opposant à l'information des parents des enfants hétérozygotes :
- « [Que] la révélation systématique du statut de porteur sain d'un nouveau-né ne soit pas encouragée, compte tenu de l'absence d'intérêt direct pour l'enfant. »
- « Il s'agit de ne pas transformer un être humain en un être enfermé dans son statut génétique, avec le risque de sacralisation du gène que cela comporte. »
L'information concernant l'hétérozygotie ne devrait être délivrée que sur une demande de l'enfant dépisté et arrivé en âge de procréer mais sachant que ces enfants hétérozygotes et leurs parents sont convoqués pour un test de la sueur, il est difficile de ne pas révéler cette information dès la naissance. De même, les parents d'enfants considérés comme hétérozygotes, sont en droit de s'interroger sur leur propre hétérozygotie notamment dans le cas où ils auraient l'intention d'avoir des enfants dans le cadre d'une autre union.
Dépistage prénatal
Un diagnostic anténatal ou pré-implantatoire est proposé si le conseil génétique détermine qu'un couple a un risque de 25 ou de 50 % d'avoir un enfant malade, ce qui est le cas chez les couples hétérozygotes déjà détectés lors de la naissance d'un enfant malade. Les demandes de couples à faible risque (fratrie de parents d'enfants malades) sont également de plus en plus fréquentes.
Chez les couples hétérozygotes, l'analyse de l'ADN du fœtus pour recherche de mutation, par biopsie de villosité choriale peut être réalisé de façon fiable et précoce à 10 semaines d'aménorrhée . Si cette analyse moléculaire est impossible on peut pratiquer une amniocentèse à 18 semaines pour le dosage des isoenzymes de la phosphatase alcaline dans le liquide amniotique. Un taux normal élimine une mucoviscidose alors qu'un taux effondré est évocateur mais non spécifique et nécessite la réalisation d'un caryotype.
Une obstruction intestinale fœtale découverte de façon fortuite lors d'une échographie doit faire rechercher la mucoviscidose dont le risque est alors estimé entre 2 et 5 %. Cette anomalie peut être révélée par une hyperéchogénicité ou une dilatation intestinale et elle est secondaire à diverses affections telles qu'un illéus méconial, une atrésie jéjunoiléales, un syndrome de bouchon méconial, un volvulus ou une péritonite méconiale, qui peuvent toutes révéler une mucoviscidose. L'absence de visualisation de la vésicule biliaire au cours de l'échographie fœtale est aussi considérée comme un signe d'appel. Dans ces cas, un diagnostic prénatal est proposé aux parents.
Le dépistage anténatal systématique à toutes les femmes enceintes n'a pas été recommandé par le Comité consultatif national d'éthique pour les sciences de la vie et de la santé (CCNE) dans son avis n° 83 du 25 mars 2004 qui avait conclu que « le dépistage prénatal est entièrement justifié en cas d'antécédents familiaux ou de connaissance d'hétérozygotie chez un membres du couple, qu'il est à encourager au stade prénuptial ou préconceptionnel pour des familles à risque, qu'il peut trouver une légitimité dans les régions à prévalence importante du gène muté, mais qu'en revanche sa généralisation à l'ensemble d'une population pose des problèmes non seulement éthiques, mais aussi scientifiques, juridiques et économiques. »
Pour les grossesses à haut risque de transmission de la maladie, le diagnostic préimplantatoire est possible chez les couples devant recourir à une procréation médicalement assistée et chez qui la mutation a été identifiée. Ce diagnostic autorisé en France et aux États-Unis mais pas dans tous les pays, consiste à faire l'étude de l'ADN recueilli sur des cellules de l'embryon obtenu après fécondation in vitro. Seuls les embryons non porteurs de la mutation ou hétérozygotes sont transférés.
Conseil génétique
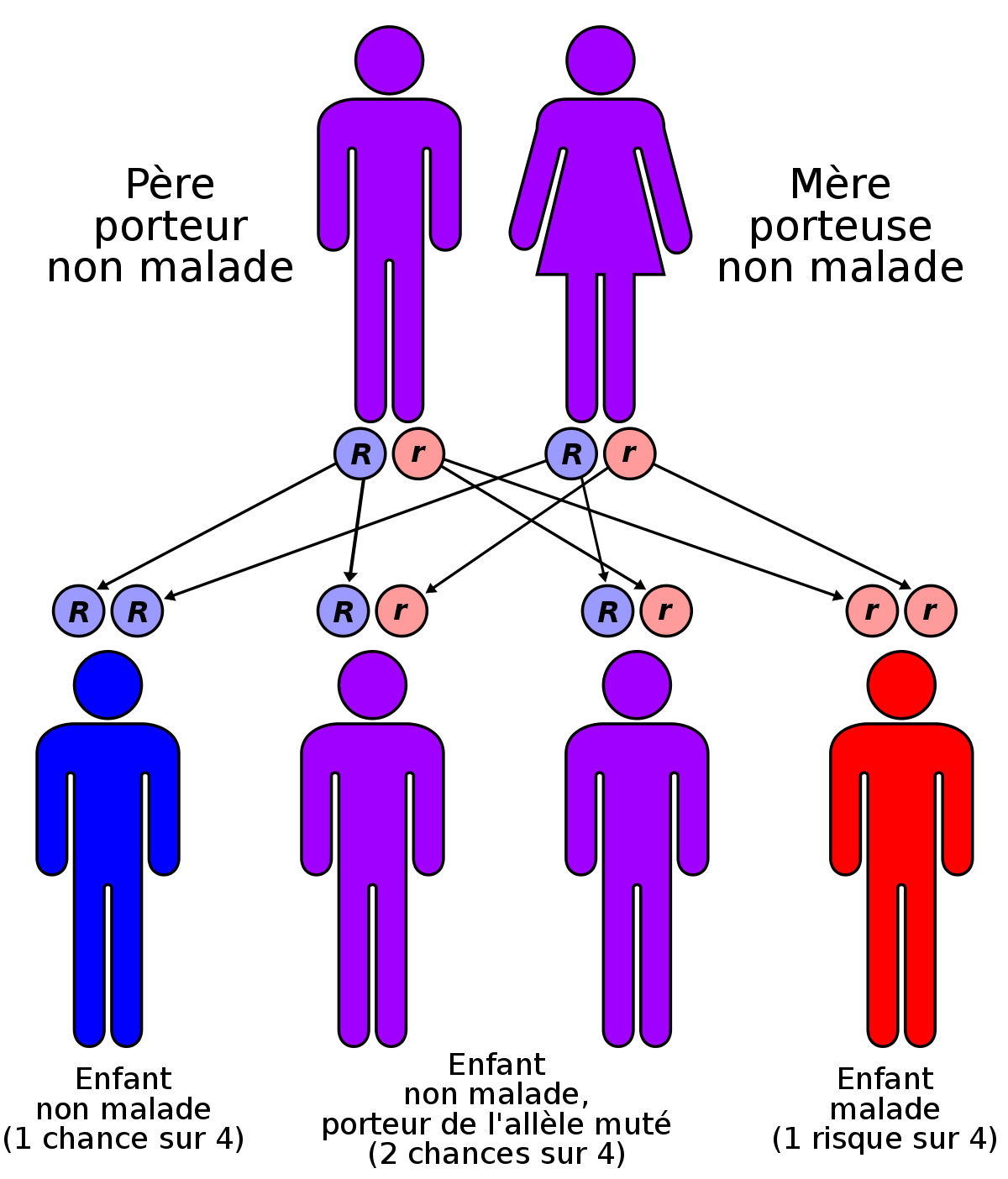
Le conseil génétique a pour but d'apprécier le risque de récurrence - d'apparition - de la maladie au sein d'une famille à risque. Il découle du mode de transmission de la maladie qui est récessive et non liée au sexe ce qui implique que le risque de récurrence est de 1 sur 4, c'est-à-dire que si deux parents sont porteurs hétérozygotes d'une mutation pathogène du gène CFTR, ils ont un risque de 25 % d'avoir un enfant homozygote malade. Ils ont également une chance sur 4 d'avoir un enfant sain porteur du gène normal et une chance sur 2 d'avoir un enfant hétérozygote, porteur sain d'une seule allèle anormale et non malade.
Risque pour les membres d'une famille d'un enfant mucoviscidosique
- Frères et sœurs d'un enfant malade
La probabilité d’être hétérozygote pour les frères et sœurs d'un malade est de 2 sur 3. Une recherche de mutation est possible afin de préciser le statut génétique de chaque membre de la fratrie. Cette recherche est importante dans le cadre d’un projet parental; si un porteur sain est détecté, son conjoint peut également bénéficier d'une recherche de mutations afin de déterminer si un risque existe.
- Descendant d'un sujet atteint de mucoviscidose
Les femmes sont fertiles, mais il leur est difficile d'avoir un enfant. Les hommes sont stériles à cause de l'atrésie bilatérale des canaux déférents et devront avoir recours aux techniques de procréation médicalement assistée. Chacun des descendants d'un couple dont un des membres est atteint de la mucoviscidose sera porteur d'un gène de la maladie, le risque d'être malade - d'être homozygote - dépendra du statut génétique du conjoint du patient; si celui-ci n'est porteur d'aucune mutation, le risque est très faible (mais possible dans le cas d'une mutation rare non détectée); s'il est hétérozygote, le risque sera de 1 sur 2. Le dépistage des mutations du CFTR chez le conjoint dépendra de son histoire familiale et de ses convictions religieuses et personnelles.
Dépistage des porteurs hétérozygotes
Pour que l'enfant présente la maladie, il faut que les deux parents soient porteurs sains hétérozygotes d'une mutation du gène CFTR (ou plus rarement qu'un parent soit atteint de la maladie et l'autre porteur sain hétérozygote). Environ deux millions de personnes sont porteur sain d'une mutation du gène CFTR.
Si les deux parents sont porteurs sains d'une mutation du gène alors l'enfant a une chance sur quatre de porter la maladie, si aucun des deux parents ou un seul est porteur d'une mutation du gène CFTR, il n'y a aucune chance que leurs enfants à naître ait la maladie. Le dépistage d'un porteur sain ne peut se faire que par analyse de son ADN à l'aide de test moléculaire puisqu'il ne présente pas de symptôme. Pour déterminer si les parents d'un couple sont porteurs sains, notamment à l'occasion d'une grossesse, un dépistage de la femme peut être proposé (car c'est elle qui consulte le plus souvent et en cas de test positif), un test peut être fait par la suite sur le père.
Toutefois, cette méthode ne permet pas l'éradication de la maladie. En effet, même si un test prénatal est effectué sur les deux parents, et que l'un de ces tests est négatif, cela ne garantit pas que l'enfant n'aura pas la maladie puisque l'analyse ADN du gène CFTR peut donner de faux négatifs, toutes les mutations du gène n'étant pas connues (la principale mutation ΔF508 représentant moins de 70 % des cas).
Dépistage lors du don de sperme
En France, les généticiens proposent de ne pas dépister systématiquement les donneurs hétérozygotes pour rester dans la même situation qu'en procréation naturelle où le dépistage systématique n'est pas justifié.

















































