Mucoviscidose - Définition
La liste des auteurs de cet article est disponible ici.
Étiologie et physiopathologie
Le gène responsable de la mucoviscidose

Le gène CFTR, Cystic fibrosis transmembrane conductance regulator, code la synthèse d'une protéine appelée CFTR dont la fonction est essentielle à l'organisme. Les mutations dans ce gène sont responsables de la mucoviscidose et de l'aplasie congénitale du canal déférent. C'est un grand gène constitué d'environ 250 000 paires de bases répartis en 27 exons et codant un ARNm de 6,5 kb. Il est localisé sur le locus 7q31.2, dans la région q31.2 du bras long du chromosome 7.
Mutations du gène CFTR
On dénombre en 2007 plus de 1 500 mutations du gène CFTR.
La majorité des mutations du gène CFTR sont des mutations ponctuelles. Les plus fréquentes sont des mutations faux-sens (42%), puis viennent des microinsertions et des microdélétions (24%), des mutations non-sens (16%), des mutations d'épissage (16%) et enfin des délétions d'un acide aminé (2%). On rapporte également quelques grandes délétions.
Les mutations du gène CFTR sont regroupées en 6 classes en fonction des conséquences fonctionnelles qu'elles occasionnent. Certaines mutations entraînent des anomalies quantitatives ou qualitatives sur la protéine CFTR:
- Classe 1 : mutations altérant la production de la protéine CFTR.
- Classe 2 : mutations perturbant le processus de maturation cellulaire de la protéine CFTR.
- Classe 3 : mutations perturbant la régulation du canal chlorure.
- Classe 4 : mutations altérant la conduction du canal chlorure.
- Classe 5 : mutations altérant la stabilité de l'ARNm CFTR.
- Classe 6 : mutations altérant la stabilité de la protéine mature.
La mutation la plus fréquente est la mutation Delta F508 (ΔF508) qui consiste en une délétion de trois nucléotides au niveau du dixième exon du gène, aboutissant à l'élimination d'un acide aminé, la phénylalanine, en position 508. Cette mutation est retrouvée dans près de deux tiers des cas avec des variations importantes selon les populations étudiées, ainsi on retrouve un gradient de répartition suivant une direction nord-ouest/sud-est en Europe, avec par exemple 88% de DF508 au Danemark, 81% en Bretagne et 50% en Italie. Seule quatre autres mutations, hors ΔF508, représentent plus de 1% des cas, il s'agit de G542X, G551D, N1303K et W1282X. Toutes les autres mutations sont rares, voire exceptionnelles, uniquement retrouvées au sein d'une seule famille.
Les mutations les plus fréquentes dans les populations caucasiennes sont :
| Nom de la mutation | Fréquence | Nom de la mutation | Fréquence | |
|---|---|---|---|---|
| ΔF508 | 66.0 % | R553X | 0.7 % | |
| G542X | 2.4 % | 621+1G | 0.7 % | |
| G551D | 1.6 % | 1717-1G | 0.6 % | |
| N1303K | 1.3 % | R117H | 0.3 % | |
| W1282X | 1.2 % | R1162X | 0.33 % |
Les génotypes retrouvés sont:
| Paire de mutations | Fréquence |
|---|---|
| ΔF508 - ΔF508 (homozygote) | 50% |
| ΔF508 - autre mutation (hétérozygote composite) | 40% |
| autre mutation - autre mutation | 10% |
Corrélation génotype et phénotype
Il existe un certain rapport entre la mutation (le génotype) et les manifestations cliniques de la maladie (le phénotype). La très grande variabilité des manifestations et de la gravité de cette maladie est en rapport avec les multiples mutations du gène CFTR.
La pénétrance est habituellement de 100% chez les homozygotes avec mutations sévères mais la sévérité de la maladie reste variable avec des formes atténuées sans insuffisance pancréatique ou avec atteinte respiratoire modérée.
L'état homozygote ΔF508/ΔF508 est associée à la forme classique de la maladie avec une augmentation des électrolytes dans la sueur, une insuffisance pancréatique et une atteinte pulmonaire souvent sévère. Certaines mutations entraînent plus de troubles fonctionnels que d'autres ainsi les études épidémiologiques montrent une mortalité différente dans des populations ayant des mutations différentes. Ainsi les mutations ΔF508/R117H, ΔF508/DeltaI507, ΔF508/3849+10kbC-→T et ΔF508/2789+5G-→A ont un taux de mortalité plus bas que les homozygotes DeltaF508. Et les mutations ΔF508/R117H, ΔF508/DeltaI507, ΔF508/ 3849+10 kbC-→T, ΔF508/2789+5G-→A et ΔF508/A455E ont des manifestations phénotypiques atténuées.
C'est pour la fonction pancréatique que la corrélation génotype/phénotype est la plus évidente, le pancréas peut présenter des lésions de gravité très variable selon les mutations, alors que la sévérité de l'atteinte pulmonaire peut être très différente chez des sujets ayant un même génotype CFTR, même au sein d'une même famille. Cette variabilité phénotypique de l'atteinte respiratoire pourrait être due à des facteurs génétiques distincts du gène CFTR ou à des facteurs environnementaux. Ainsi les hétérozygotes ΔF508/A455E ont une meilleure fonction pulmonaire que les homozygotes ΔF508; cette mutation étant associée à une fonction pancréatique conservée, la moindre sévérité de l'atteinte pulmonaire pourrait être la conséquence d'un meilleur état nutritionnel.
La protéine CFTR
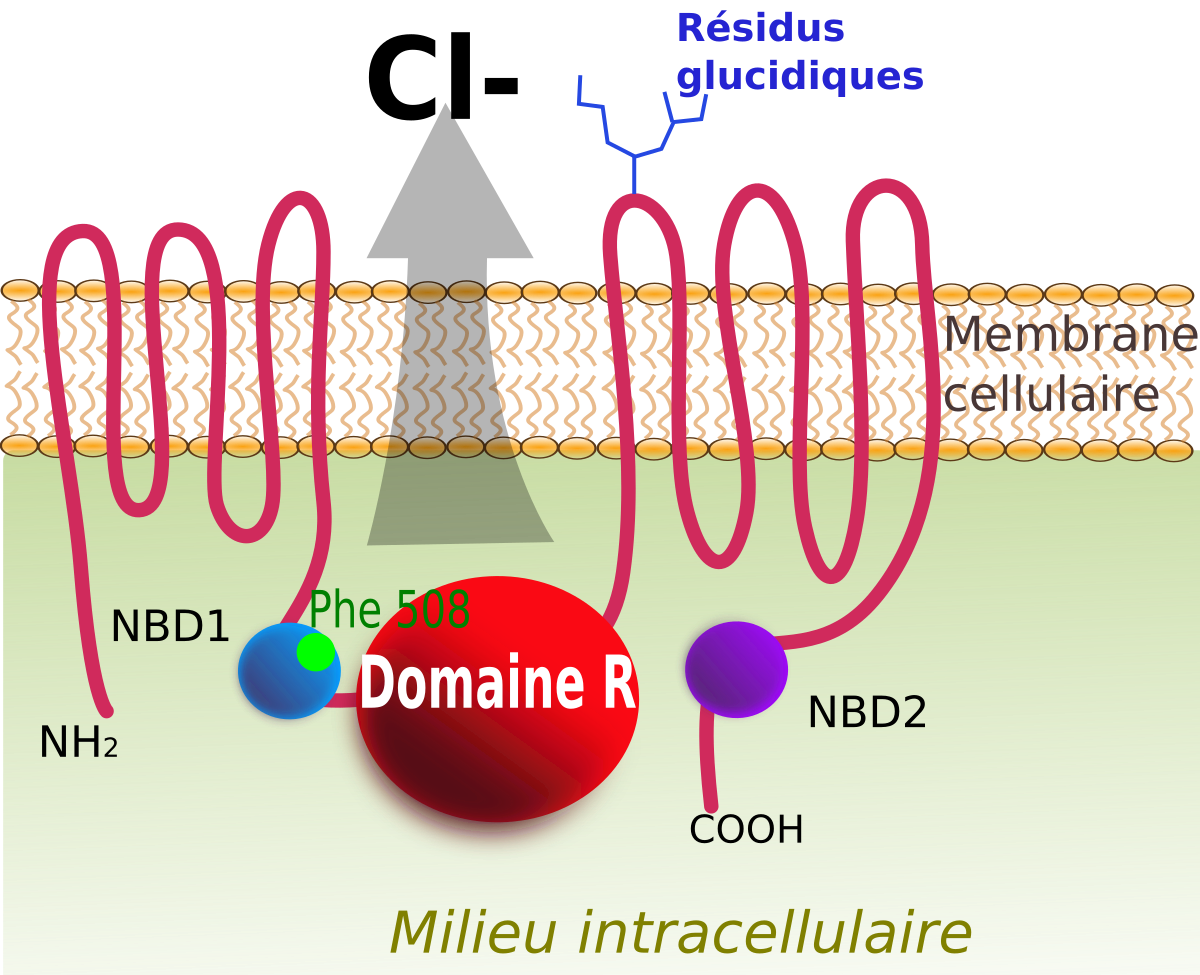
La protéine CFTR (cystic fibrosis transmembrane conductance regulator) est codée par le gène CFTR. C'est une protéine membranaire de 1 480 acides aminés et son poids moléculaire est de 168 kD.
La molécule CFTR est composée de 5 domaines, deux domaines hydrophobe transmembranaires (membrane-spanning domains, MSD) comportant chacun six segments transmembranaire en hélice alpha, deux domaines hydrophile d'interaction avec les nucléotides (nucleotide-binding domains, NBD), et un domaine cytoplasmique de régulation (regulatory domain, R) codé par l'exon 13 et contenant de nombreux résidus chargés et la majorité des sites potentiels de phosphorylation.
La molécule se situe au pôle apical des cellules épithéliales et se comporte comme un canal ionique laissant passer l'ion chlorure, thiocyanate selon un gradient électrochimique (transport actif secondaire, tirant son énergie du gradient électrochimique favorable à l'entrée de l'ion sodium dans la cellule entretenu par l'activité de la pompe sodium/potassium). L'activation du canal dépend de multiples phénomènes de phosphorylation du domaine de régulation (R) et de l'hydrolyse d'une molécule d'Adénosine triphosphate (ATP) sur les domaines d'interaction avec les nucléotides. L'hydrolyse du NBD en situation N-terminal permet l'ouverture du canal transmembranaire sélectif aux ions chlorure. L'hydrolyse de l'ATP sur le domaine NBD C-terminal, conduit à sa fermeture.
Outre sa fonction canal chlore, la protéine CFTR régule également d'autres canaux comme le canal chlore à rectification sortante, le canal sodium épithélial, et des canaux potassium à rectification entrante. D'autres fonctions sans rapport avec la régulation des canaux ont aussi été décrites comme le transport d'ATP, la modulation des phénomènes d'exocytose et endocytose, la régulation du pH des organelles intracellulaires. Ce qui fait de la protéine CFTR une protéine multi-fonctionnelle.
Physiopathologie des manifestations
La mutation du gène de la mucoviscidose entraîne un défaut dans la synthèse de la protéine cystic fibrosis transmenbrane conductance regulator ou CFTR (famille ATP Binding Cassette), qui est une protéine membranaire entrant dans la formation d'un canal ionique sélectif aux anions et principalement chlorures.
Le défaut de synthèse de la protéine entraîne un déficit en chlore extracellulaire, en thiocyanate et donc un défaut d'hydratation du mucus, une hyperviscosité des sécrétions épithéliales liée à une réabsorption exagérée de l'eau secondaire à la rétention cellulaire de l'ion chlore et au non passage des composés nécessaires à la défense immmunitaire du poumon. Ainsi, le composé hypothiocyanite ne peut être produit du fait du déficit en thiocyanate n'étant pas transféré par le CFTR , les protéines duox étant quant à elles, inhibées par les Pseudomonas aeruginosa, approche validée par les deux agences médicales Européenne et Américaine
L'insuffisance de fonctionnement des glandes exocrines se remarque surtout au niveau du poumon, du pancréas et du foie. Les atteintes digestives sont les plus précoces et les premières historiquement décrites.
Au niveau du pancréas
Les enzymes du pancréas, comme la lipase et la trypsine, qui servent à la digestion sont mal excrétées dans la lumière intestinale parce que les canaux pancréatiques se bouchent du fait d'une sécrétion trop épaisse et d'une fibrose importante. Les enzymes agressent alors directement le tissu pancréatique, contribuant à la fibrose. Des kystes peuvent éventuellement se former. Lorsque les fonctions pancréatiques sont significativement altérées, on parle d'insuffisance pancréatique.
Le pancréas est atteint dans sa fonction exocrine et endocrine de deux manières :
- atteinte de la fonction exocrine : la réduction ou l'absence d'arrivée d'enzymes de digestion dans la lumière intestinale conduit à un syndrome de malabsorption pouvant avoir un retentissement sur le développement staturo-pondéral et pubertaire des enfants.
- atteinte de la fonction endocrine : la destruction des cellules bêta des îlots de Langerhans conduit à un défaut de sécrétion d'insuline et un diabète insulinoprive - mais bien distinct du diabète de type 1 auto-immun - apparait. Si l'insuffisance pancréatique exocrine apparaît précocement, les îlots de Langerhans sont longtemps épargnés. Il semble que la destruction des îlots ne soit pas causée uniquement par le développement progressif d'une fibrose pancréatique mais aussi par d'autres phénomènes tels qu'une ischémie. La capacité à sécréter de l’insuline reste longtemps normale et plusieurs plusieurs années s'écoulent entre la période d'intolérance au glucose et l'apparition du diabète. Outre les anomalies de sécrétion de l'insuline, d'autres facteurs pourraient contribuer développement du diabète tels qu'une insulino-résistance transitoire.
Au niveau hépatobiliaire
La viscosité de la bile est augmentée par les dysfonctionnements de la protéine CFTR des cellules épithéliales biliaires. Les canaux qui transportent la bile vont se boucher à cause de ce liquide trop épais. La répétition de ces obstructions conduit à des phénomènes de cirrhose localisée et d'hépatomégalie.
Les conséquences habituelles de la cirrhose biliaire sont les mêmes chez le patient mucoviscidosique que dans le reste de la population. L'hypertension dans la veine porte, les hémorragies digestives et l'insuffisance hépatocellulaire peuvent conduire à la greffe hépatique.
Au niveau du poumon
Le liquide de surface tapissant l'arbre bronchique se compose d'eau et de mucus. Les canaux CFTR servent à la sécrétion active de chlore vers ce liquide, ils interagissent avec les canaux d'absorption du sodium pour limiter leur travail. Ce mouvement de chlore entraîne un mouvement de sodium et d'eau. Cette sécrétion d'eau permet d'hydrater le liquide de surface bronchique et de lui maintenir des propriétés rhéologiques adéquates pour une clairance muco-ciliaire efficace. La clairance muco-ciliaire est le débit de liquide de surface transporté par les cellules ciliées de l'arbre bronchique. L'évacuation de ce liquide permet l'élimination des poussières et des agents infectieux vers le système digestif.
L'altération fonctionnelle des canaux CFTR entraîne une déshydratation du liquide de surface bronchique. Les modifications des propriétés du mucus, et notamment l'augmentation de sa viscosité rendant plus difficile son évacuation par les cils, conduit à une obstruction chronique des bronches ainsi qu'à la non évacuation des poussières et bactéries. De plus les propriétés antibactériennes du mucus sont diminuées. Tous ces éléments favorisent l'apparition d'une infection précoce devenant rapidement chronique associée à une réaction inflammatoire marquée.
Au fil du temps, différentes bactéries colonisent les voies respiratoire d'un même malade. Une large étude publiée en 2000 faite sur 1 000 enfants de moins de 2 ans retrouvait sur des prélèvements bronchoscopiques 19 % d'Haemophilus influenzae, 42 % de Staphylococcus aureus, 29 % de Pseudomonas aeruginosa, 7 % de Stenotrophomonas maltophilia et moins de 1 % de Burkholderia cepacia. D’autres agents infectieux, bactériens, viraux ou fongiques, peuvent parfois être retrouvés. Initialement ce sont des bactéries banales telles que Haemophilus influenzae et Staphylococcus aureus qui colonisent et infectent les poumons, puis survient la colonisation à Pseudomonas aeruginosa, qui constitue un tournant dans l’évolution de la maladie respiratoire. L'utilisation répétée d'antibiotiques mais aussi d'autres facteurs encore indéterminés et liées à la maladie elle-même, favorisent la colonisation du tractus bronchique par des bacilles pyocyaniques généralement résistants aux antibiotiques d'utilisation courante. Ces bactéries sont exceptionnelles chez les personnes indemnes de mucoviscidose.
Il existe une inflammation de l'épithélium bronchique dont les mécanismes sont incomplètement élucidés. Il n'est pas encore tranché que cette inflammation soit directement consécutive à la colonisation bactérienne du mucus. Les études physiopathologiques se concentrent sur les autres fonctions de la protéine CFTR, qui aurait un rôle dans la régulation de l'inflammation, voire un rôle dans la destruction de Pseudomonas Aeruginosa.
À terme, l'inflammation et l'infection chronique entretiennent un cercle vicieux et entraînent une dégradation pulmonaire par des lésions du tissu pulmonaire conduisant à un tableau de broncho-pneumopathie chronique obstructive.

















































