Maladie du sommeil - Définition
La liste des auteurs de cet article est disponible ici.
Cycle parasitaire
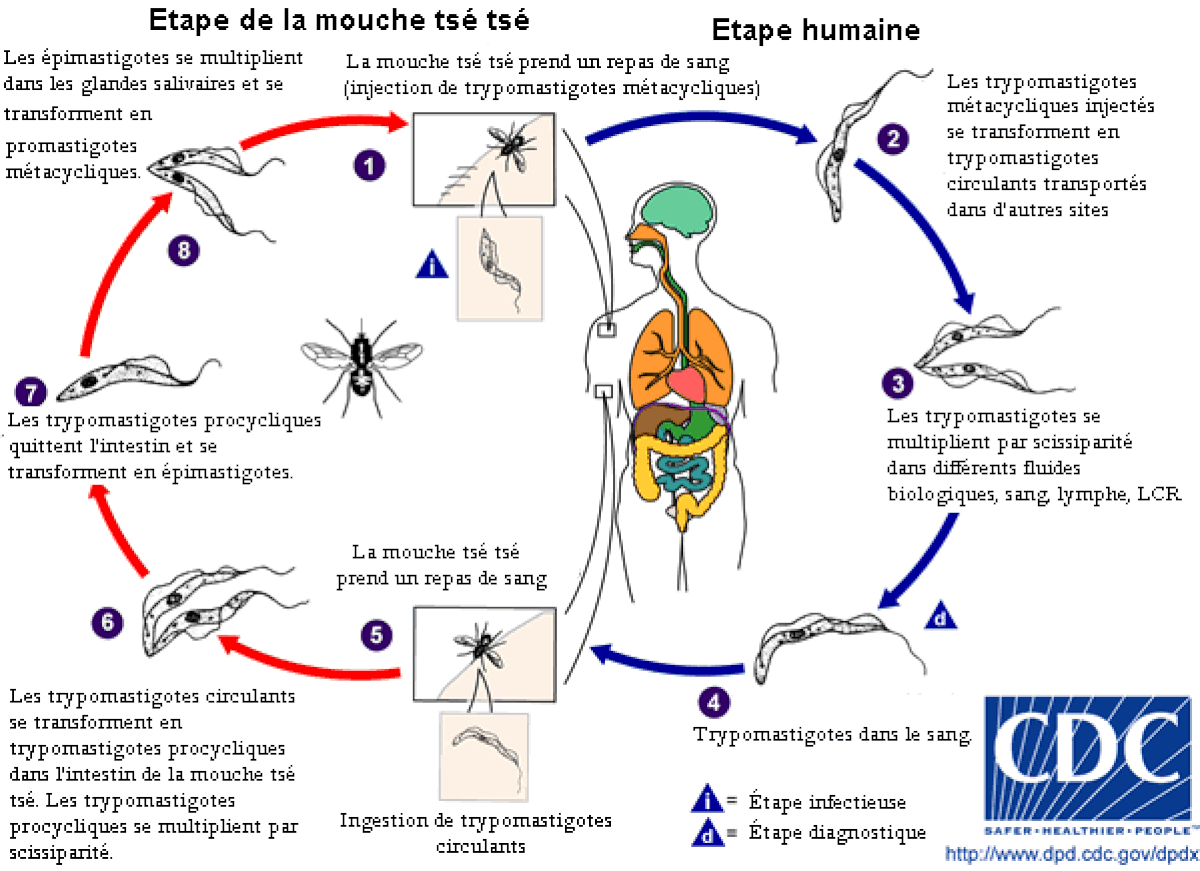
La mouche tsé-tsé est grande, brune et furtive. La piqûre est ressentie comme une aiguille chaude enfoncée dans la chair. Pendant un repas de sang sur le mammifère hôte, une mouche tsé-tsé infectée du (genre Glossina ) injecte les trypomastigotes métacycliques dans le tissu cutané. Les parasites entrent dans le système lymphatique et passent dans la circulation sanguine (1). À l'intérieur de l’hôte, ils se transforment en trypomastigotes circulants dans le sang (2) et sont transportés à d'autres emplacements dans tout le corps, atteignent d'autres fluides biologiques (par exemple, lymphe, liquide céphalo-rachidien), et continuent de se répliquer par scissiparité (3). Le cycle parasitaire du Trypanosome africain est représenté par des étapes extracellulaires. Une mouche tsé-tsé s’infecte par des trypomastigotes circulants en prenant un repas de sang sur un mammifère infecté hôte (4)(5). (Dans l’intestin de la mouche, les parasites se transforment en trypomastigotes procycliques, se multiplient par scissiparité (6), quittent l’intestin, et se transforment en épimastigotes (7). Les épimastigotes atteignent les glandes salivaires de la mouche et continuent leur multiplication par scissiparité (8). Le cycle dans la mouche dure approximativement 3 semaines.
Prévention et contrôle
Il n'existe pas de vaccin ni de prévention médicamenteuse.
La prévention et le contrôle se focalisent, là où c’est possible, sur l'extirpation de l'hôte parasite, la mouche tsé-tsé. Deux stratégies ont été employées alternativement dans les tentatives pour réduire les trypanosomiases africaines. L'une des tactiques est principalement médicale ou vétérinaire et vise directement la maladie en utilisant la prophylaxie, le traitement, et la surveillance pour réduire le nombre d'organismes porteurs de la maladie. La deuxième stratégie est généralement entomologique et prévoit de perturber le cycle de transmission en réduisant le nombre de mouches.
Il existe des exemples de réduction de la maladie du sommeil par l'utilisation de techniques de stérilisation des insectes.
La surveillance active régulière, impliquant la détection et le traitement des cas, en plus du contrôle des mouches de tsé-tsé, est l'épine dorsale de la stratégie pour le contrôle de la maladie du sommeil. Le dépistage systématique dans les communautés où des foyers ont été identifiés est la meilleure approche, car le dépistage au cas par cas n'est pas possible en pratique dans les zones de forte endémie. Le dépistage systématique peut se faire sous forme de cliniques mobiles ou de centres fixes de dépistages où les équipes voyagent quotidiennement dans les foyers. La nature de la maladie de gambiense est telle que les patients ne cherchent pas à se faire soigner assez tôt parce que les symptômes à cette phase ne sont pas évidents ou assez sérieux pour justifier une consultation médicale, en raison de l'éloignement des quelques secteurs affectés. En outre, le diagnostic de la maladie est difficile et la plupart des personnels sanitaires ne sont pas capable de la détecter. Le dépistage systématique permet à la maladie débutante d'être détectée et traitée avant que la maladie progresse, et diminue le réservoir humain potentiel.
Pour le voyageur, le port de vêtements couvrants ainsi que de repellents contre la mouche responsable est conseillé.
Traitement
Le traitement standard courant pour la première étape de la maladie est :
- La pentamidine en intraveineuse (pour le T.b. gambiense ) ou en intramusculaire, sur une semaine, les effets secondaires principaux étant l'hypoglycémie et la douleur au point d'injection ;
- La suramine en intraveineuse (pour le T.b.rhodesiense ) sur une durée plus prolongée.
Le traitement standard courant pour la deuxième étape de la maladie (phase neurologique) est :
- Le mélarsoprol en Intraveineuse 2.2 mg/kg et par jour pendant 10 jours consécutifs.
La première ligne de thérapies alternative inclut :
- Le mélarsoprol en intraveineuse 0.6 mg/kg le jour 1, le mélarsoprol IV 1.2 mg/kg sur le jour 2, et 1.2/jour IV combiné avec 7.5 mg/kg par voie orale de nifurtimox deux fois par jour les jours 3 à 10;
ou
- L' eflornithine en Intraveineuse 50 mg/kg toutes les six heures pendant 14 jours.
Dans les zones résistance au mélarsoprol ou chez les patients qui ont rechuté après monothérapie au mélarsoprol, le traitement devrait être :
- mélarsoprol et nifurtimox, ou
- eflornithine
Les protocoles traditionnels suivants ne devraient plus être employés :
- Ancien « schéma » thérapeutique de 26 jours de mélarsoprol (3 séries de 3.6 mg/kg/jour en intraveineuse pendant 3 jours, avec une interruption de sept jours entre les séries) (ce protocole est moins facile et les patients sont moins volontaires pour accomplir le traitement complet);
- Traitement à doses progressives de mélarsoprol : traitement de dix jours de mélarsoprol ( 0.6 mg/kg IV le jour 1, 1.2 mg/kg IV le jour 2, et 1.8 mg/kg les jours 3 à 10). Ce protocole était censé réduire le risque d'encéphalopathie induite par le traitement, mais maintenant on sait qu’il est associé à un plus grand risque de rechute et à une incidence plus élevée de l'encéphalopathie;
Tous les patients devraient être suivis pendant deux années avec des ponctions lombaires semestrielles pour détecter les rechutes.
Histoire du traitement de la maladie du sommeil
En 1910, l’année même de la découverte de Bruce, est introduit le premier médicament actif contre la maladie du sommeil, le Salvarsan (arsphénamine), mis au point par Paul Ehrlich et Sahachiro Hata. Cependant, l’emploi de ce dérivé de l’arsenic présente des risques graves, dont la cécité. Jointe à une toxicité qui le rend peu maniable, l’efficacité du Salvarsan incite à poursuivre les recherches en direction des dérivés de l’arsenic, et d'autres molécules seront obtenues par dérivation de l'acide aminophénolarsinieux : la tryparsamide et le mélarsen, acides arsiniques, à noyau d'arsenic pentavalent ; l'arséno-phénylglycine, le melarsen-oxyde et le melarsoprol, oxydes d'acides arsiniques, à noyau trivalent.
Il faut pourtant attendre jusqu’en 1919 pour qu’aboutissent les travaux de Walter Jacobs et Michael Heidelberger, qui permettront à Wade Brown et Louise Pearce d’introduire la tryparsamide. Dès lors, trois trypanocides apparus au cours des années 1920, la suramine (non-arsenical), l’orsanine et la tryparsamide, sont employés dans de vastes campagnes de dépistage et de traitement de masse. La suramine (Bayer 205), synthétisée en 1917, mais dont la formule, tenue secrète, n'est élucidée par Ernest Fourneau qu'en 1924 à l'Institut Pasteur, est introduite dès 1920. Elle est utilisée au premier stade de la maladie ou en cas d'arséno-résistance. Son administration par voie intraveineuse et à des doses élevées en limite l’emploi. Cependant, et malgré son importante toxicité, elle est encore en usage en 2001 dans la phase lymphatico-sanguine. La tryparsamide, dérivée de l'atoxyl, est introduite en 1921 et commercialisée en 1930 sous le nom de Tryponarsil. C’est le premier médicament actif sur la phase encéphalo-rachidienne. Cependant, même réduite par une posologie progressive, sa toxicité oculaire touche 2 % des malades traités au cours de la campagne menée par Jean Laigret au Congo Brazzaville au milieu des années 1920, et 4,4 % de ceux traités par Eugène Jamot au Cameroun à la même époque. Et chez les patients administrés à un stade avancé, le taux de létalité iatrogène s’élève à 6,6 %. L'orsanine sodique (Fourneau 270) est employée à partir de 1925 et pendant une quinzaine d'années, soit seule chez les patients « en période douteuse », soit en association avec la tryparsamide chez les malades au stade second.
Dès le début des années 1930, au Congo belge, apparaissent les premiers cas d’arséno-résistance et, à partir de 1934, la tryparsamide est associée à d'autres molécules, dont la suramine et l'orsanine sodique.
L’orsanine est abandonnée à la découverte, en 1939, de la pentamidine, molécule très efficace dans la première phase de la maladie, couramment employée en Afrique occidentale comme agent prophylactique, et qui permet pendant les années 1950 une diminution des taux d'infection si importante qu'on croit possible l'éradication de la maladie. La suramine quant à elle, généralement associée à la tryparsamide, reste le médicament le plus employé dans le traitement de la maladie du sommeil jusqu’à la fin des années 1960, progressivement remplacée à partir de 1949 par le mélarsoprol, mis au point par Friedheim et qui présente une toxicité moins élevée que tous les arsenicaux employés jusqu’alors.
L’éflornithine enfin (difluoromethylornithine ou DFMO), le traitement le plus moderne, a été mise au point dans les années 1970 par Albert Sjoerdsmanot et elle a fait l’objet d’essais cliniques dans les années 1980. La molécule a été autorisée aux États-Unis par la Food and Drug Administration en 1990, mais Aventis, le laboratoire pharmaceutique responsable de sa fabrication, a cessé la production en 1999. En 2001, cependant, Aventis, en association avec Médecins sans frontières et l'Organisation mondiale de la santé, a signé un accord à long terme pour fabriquer et distribuer le médicament. Son efficacité est démontrée, sa tolérance est meilleure que celle du mélarsoprol. Il existe cependant, des rechutes sous ce traitement, dans un peu moins de 10% de cas, mais le plus souvent mortelles.
Des traitements combinés pourraient réduire ce risque de rechute, parmi lesquels l'association eflornithine - nifurtimox semble particulièrement prometteuse.
Le Megazol®, molécule biologiquement active avec une activité trypanocide, a fait l'objet il y a quelque années d'études scientifiques. En effet cette molécule, active sous forme de pilule (voie orale) permet d'éliminer 100 % du parasite en 2 jours (tests in vitro et in vivo chez le chimpanzé et le cochon). Cette molécule est cependant suspectée d'être cancérigène et son développement a été arrêté.
Perspectives et voies de recherche
Le génome du parasite a été décodé et plusieurs protéines ont été identifiées comme cibles potentielles pour un traitement médicamenteux. Le décodage de l'ADN a également permis de comprendre pourquoi la production d'un vaccin pour cette maladie a été si difficile. T brucei a plus de 800 gènes qui fabriquent des protéines qu’il utilise pour éviter la détection par le système immunitaire. (Berriman, et autres. , 2005)
Une équipe de recherche internationale travaillant en République démocratique du Congo, au Soudan et en Angola impliquant Immtech international et l’université de la Caroline du Nord à Chapel Hill ont mené les essais cliniques en phase I et débuté un essai en phase III en 2005 pour tester l'efficacité du premier traitement par voie orale de la maladie de sommeil, connu pour le moment sous le nom de « DB289 ».
Des résultats récents indiquent que le parasite ne peut pas survivre dans la circulation sanguine sans son flagelle. Cette découverte donne aux chercheurs un nouvel angle d’attaque pour éliminer le parasite.
Des chercheurs ont trouvé un mécanisme immunitaire qu'ils pensent utiliser pour créer de nouveaux traitements. Une protéine Hpr (haptoglobinrelated protein) découverte en 2006 et liée à des lipides du système sanguin peut capturer l'hémoglobine et la transporter vers le corps gras auxquels elles sont liées, qui contient une toxine fatale au trypanosome. Ce mécanisme pourrait être une solution immunitaire utilisant la dépendance des trypanosomes à l'hémoglobine. Lorsque cette dernière a préalablement été capturée par une protéine Hpr, les trypanosomes si fixent aussi à la particule graisseuse qui empoisonne alors le parasite.

















































