Insuffisance cardiaque canine - Définition
La liste des auteurs de cet article est disponible ici.
Introduction
L’insuffisance cardiaque représente une pathologie majeure du chien. On estime qu’elle affecte jusqu’à 10% de la population canine globale. Si cette affection reste complexe et grave, la connaissance de sa physiopathologie a énormément progressé ces dernières années, rendant son diagnostic plus facile, et permettant à de nouvelles thérapeutiques de voir le jour. Le pronostic s’en est trouvé nettement amélioré et il n’est pas rare maintenant de voir des chiens survivre plusieurs années avec une insuffisance cardiaque. Le but de cet article est de faire un point sur les connaissances actuelles sur l’insuffisance cardiaque du chien et son traitement, en tenant compte des données scientifiques publiées et accessibles et de leur pertinence, dans une optique de médecine basée sur les faits (‘evidence-based medicine’).
Physiopathologie
L’Insuffisance Cardiaque : définition
L’insuffisance cardiaque est l’incapacité du cœur à répondre aux besoins métaboliques des organes, c'est-à-dire à fournir une pression de perfusion suffisante pour assurer la diffusion de l’oxygène et des nutriments du sang vers les tissus.. Elle peut se manifester dans 2 circonstances différentes :
- les besoins des organes peuvent être considérablement augmentés et dépasser les capacités d’un cœur au fonctionnement normal. C’est le cas notamment lors d’hémorragie importante ou d’hyperthyroïdie.
- le cœur peut être défaillant et ne plus assurer un débit cardiaque suffisant. Ce cas représente la très grande majorité des insuffisances cardiaques chroniques, où une cardiopathie (congénitale ou acquise) altère la performance cardiaque.
La suite de cet article ne portera que sur les insuffisances cardiaques consécutives à une cardiopathie.
L’insuffisance cardiaque est un syndrome clinique, qu’il faut bien distinguer de la cardiopathie, affection cardiaque généralement à l’origine de l’insuffisance cardiaque. Les symptômes observés sont liés à la baisse du débit cardiaque et de la perfusion tissulaire et à l’augmentation de pression en amont du cœur (fatigabilité, toux, syncopes…) et non à la cardiopathie.
Les déterminants de la performance cardiaque
Pour répondre aux besoins métaboliques des tissus, l’appareil circulatoire doit assurer une pression de perfusion suffisante pour favoriser la diffusion de l’oxygène et des nutriments du compartiment vasculaire vers les tissus. Cette pression sanguine dépend de 2 facteurs, le débit cardiaque (quantité de sang pompée par unité de temps) et les résistances périphériques (résistance à l’écoulement du sang dans les vaisseaux et les tissus, dépendant principalement du tonus vasculaire).
Le débit cardiaque (DC, exprimé en l/mn) est conditionné par l’équation suivante :
- DC = FCxVES
où FC est la Fréquence Cardiaque (/mn) et VES (l)le Volume d’Ejection Systolique (volume de sang éjecté à chaque systole).
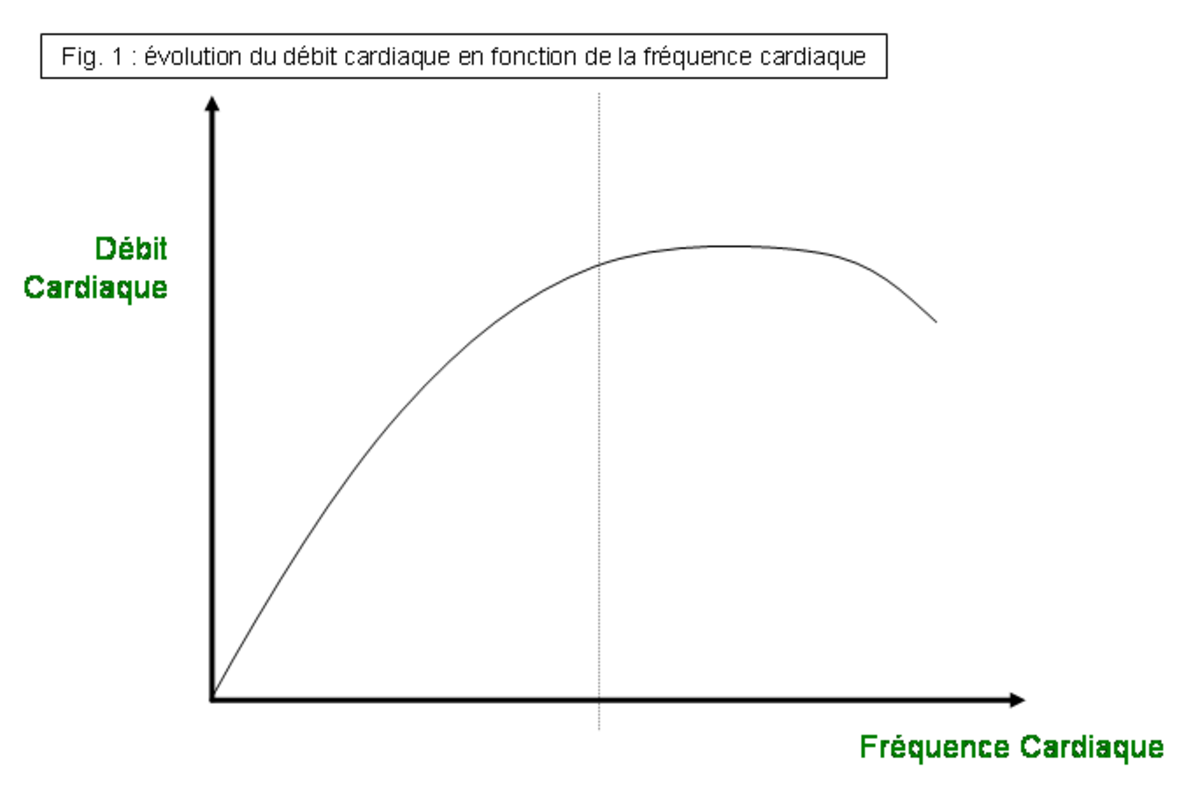
Lorsque la fréquence cardiaque augmente, le débit cardiaque augmente. Ce phénomène devient cependant autolimitant (cf. fig. 1). En effet, la durée de la systole étant pratiquement constante, une augmentation de fréquence cardiaque se fait toujours au détriment de la durée de la diastole. Lorsque la fréquence cardiaque devient excessive, le raccourcissement de la diastole entraîne une diminution du remplissage ventriculaire, ainsi qu’une baisse de l’irrigation du myocarde, à l’origine d’une baisse du volume d’éjection systolique.
Le VES est conditionné par 5 déterminants (cf. fig. 2, 3 et 4):
- 3 déterminants cardiaques :
- La contractilité = capacité des cardiomyocytes à se contracter (‘force’ de contraction du myocarde lors de la systole). Lorsque la contractilité est altérée, le VES diminue.
- La relaxation = capacité des cardiomyocytes à se relâcher (facilité de remplissage ventriculaire en diastole). Lorsque la relaxation est altérée, le VES diminue.
- La synergie de contraction = coordination du processus de contraction des chambres cardiaques, aboutissant à une éjection optimale. Lorsque la synergie de contraction est altérée (dysynergie), le processus de contraction est altéré (arythmies) et le VES diminue.
- 2 déterminants extracardiaques :
- La précharge = distension des cellules cardiomyocytes en fin de diastole, avant la contraction. Elle est conditionnée par le volume et la pression du compartiment veineux, en amont du cœur, ainsi que par le volume ventriculaire résiduel (quantité de sang restant dans le ventricule en fin de systole). Lorsque la précharge augmente, le VES augmente.
- La postcharge = résistance à l’éjection systolique. Elle dépend principalement des résistances artérielles (vasoconstriction). Lorsque la postcharge augmente, le VES diminue.
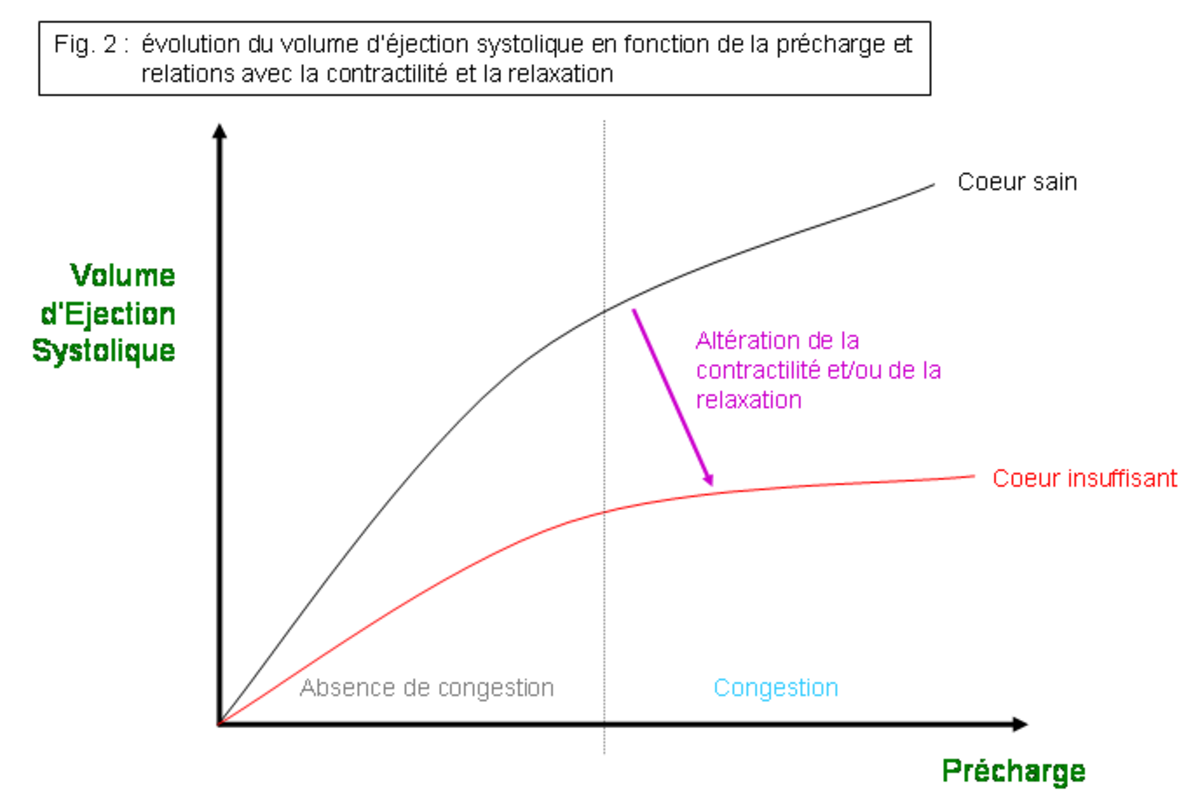
Fig. 2 : évolution du volume d’éjection systolique en fonction de la précharge et relations avec la contractilité et la relaxation | 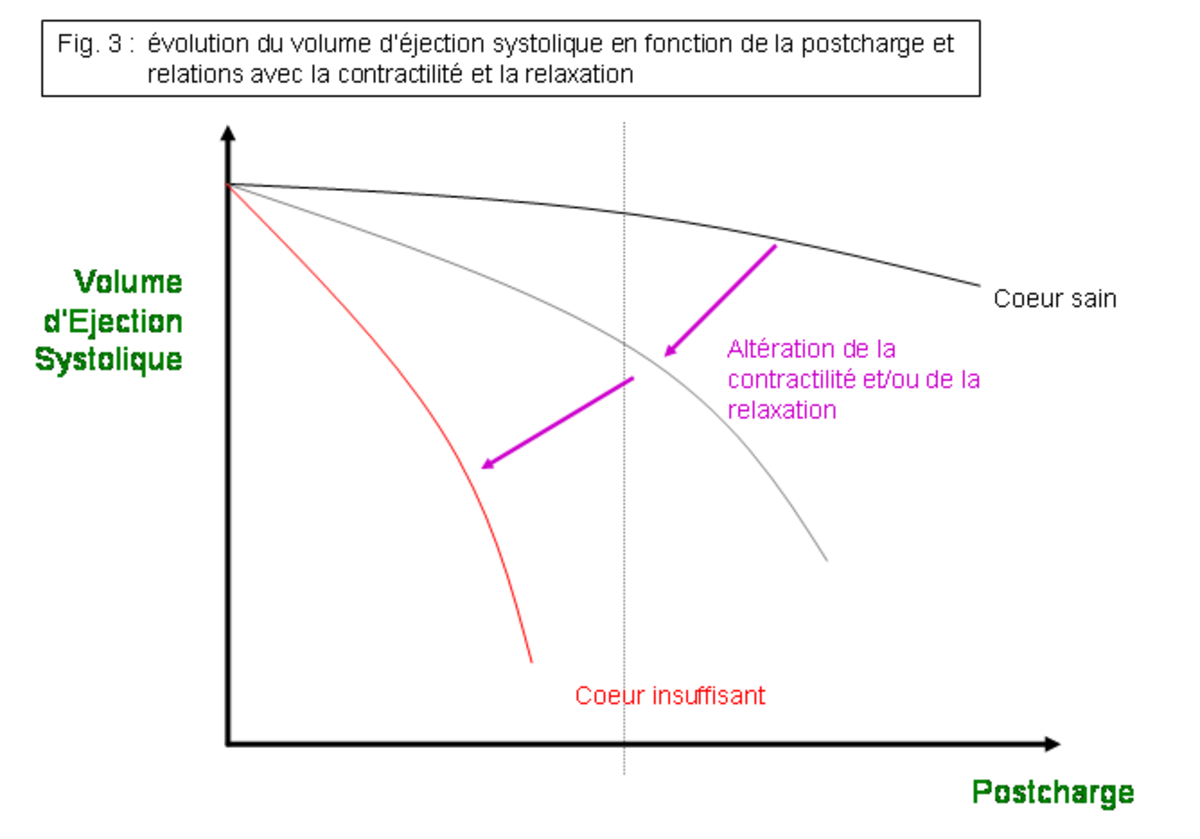
Fig. 3 : évolution du volume d’éjection systolique en fonction de la postcharge et relations avec la contractilité et la relaxation |

Les mécanismes compensateurs
Lors de cardiopathie, la performance cardiaque est altérée et la perfusion tissulaire diminuée ; l’organisme va alors mettre en place un certain nombre de mécanismes compensateurs (cf. fig. 5) afin d’en limiter les conséquences. Les deux mécanismes principaux sont :
Le système β-sympathique
La stimulation de la composante β-orthosympathique du système nerveux autonome est le premier mécanisme compensateur à se mettre en place. Elle entraîne :
- Une augmentation de la fréquence cardiaque (effet chronotrope positif) ;
- Une stimulation de la contractilité (effet inotrope positif) ;
- Une augmentation du tonus vasomoteur (effet vasoconstricteur) artériel (augmentation de la postcharge) et veineux (augmentation de la précharge) ;
Par ces mécanismes, le système β-sympathique concourt à soutenir le débit cardiaque et favoriser la perfusion tissulaire. Le β-sympathique stimule également la sécrétion de rénine par l’appareil juxtaglomérulaire (cf. ci-dessous).
Le Système Rénine Angiotensine Aldostérone (SRAA)
La baisse de perfusion rénale résultante de la baisse du débit cardiaque entraîne la sécrétion par l’appareil juxtaglomérulaire d’une hormone, la rénine. Cette hormone libérée dans le compartiment vasculaire clive l’angiotensinogène, un peptide inactif présent dans le plasma, en angiotensine I, prohormone également inactive. Dans les capillaires, l’angiotensine I rentre en contact avec une enzyme transmembranaire située à la face luminale des cellules endothéliales, l’Enzyme de Conversion de l’Angiotensine (ECA), qui la dégrade en angiotensine II, peptide vasoactif à l’origine d’une très forte activité vasoconstriction artérielle et veineuse, résultant en une augmentation de la précharge et de la postcharge. Dans un second temps, l’angiotensine II déclenche la sécrétion par la corticosurrénale d’une hormone minéralocorticoïde, l’aldostérone, qui agit sur le tube contourné distal en augmentant la réabsorption de sodium et l’élimination de potassium. L’hypertonie plasmatique résultante stimule d’une part la réabsorption d’eau et augmente ainsi la volémie (augmentation de la précharge et de la postcharge), et d’autre part contribue à activer la sécrétion de rénine, renforçant ainsi le SRAA. L’aldostérone a également une activité vasoconstrictrice directe, ainsi qu’un effet profibrotique (facteur de croissance) sur le cœur et le rein. Enfin, l’hypertonie plasmatique entraîne la libération par la neurohypophyse d’Hormone Anti-Diurétique (ADH), qui favorise la rétention d’eau et contribue ainsi à remonter la volémie et augmenter ainsi précharge et postcharge. Les effets de l’activation du SRAA sont donc une augmentation de la précharge et de la postcharge, par le biais d’une vasoconstriction et d’une hypervolémie.

La progression de l’insuffisance cardiaque
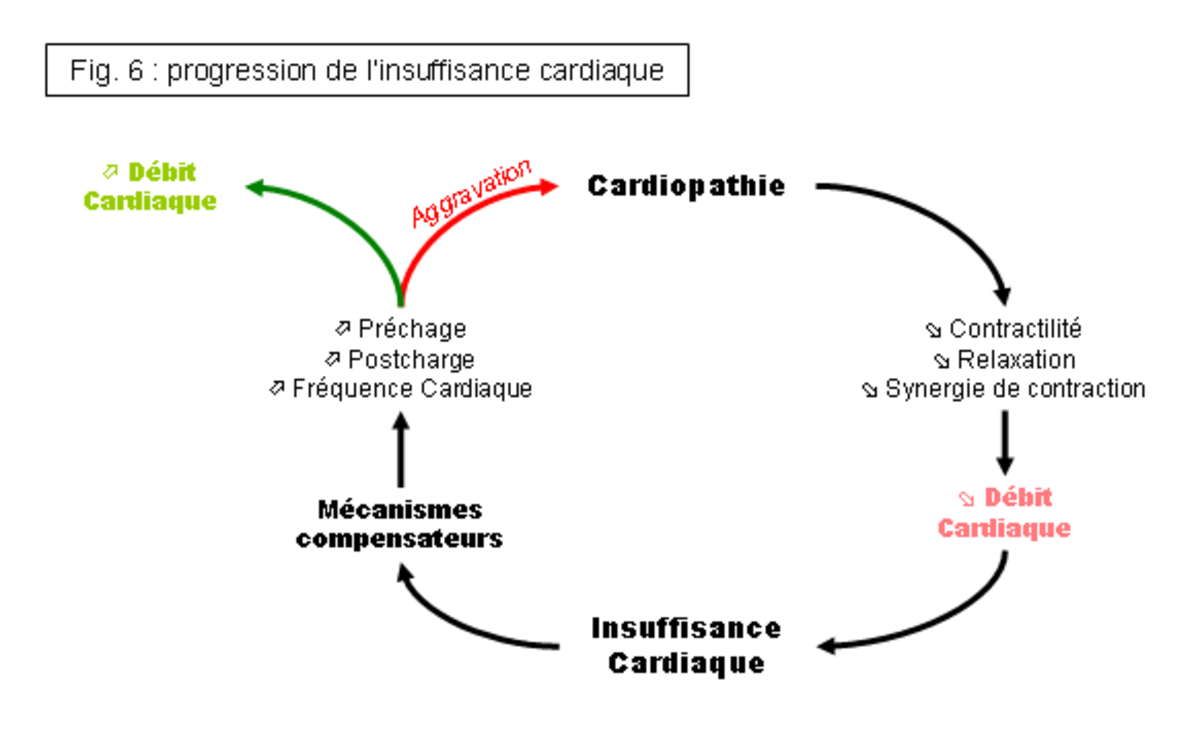
Les mécanismes compensateurs contribuent donc à soutenir le débit cardiaque (augmentation de la fréquence cardiaque, de la contractilité et de la précharge) et la perfusion tissulaire (augmentation de la postcharge) lors d’une baisse de la performance cardiaque (cardiopathie) et sont donc à court terme bénéfiques. Cependant, leur activation augmente la dépense énergétique du myocarde et ces mécanismes s’avèrent rapidement délétères à long terme, en contribuant à la progression et l’aggravation de la cardiopathie. La performance cardiaque chute alors de plus en plus, et les mécanismes compensateurs s’amplifient progressivement.
Compensation et décompensation : L’insuffisance cardiaque congestive
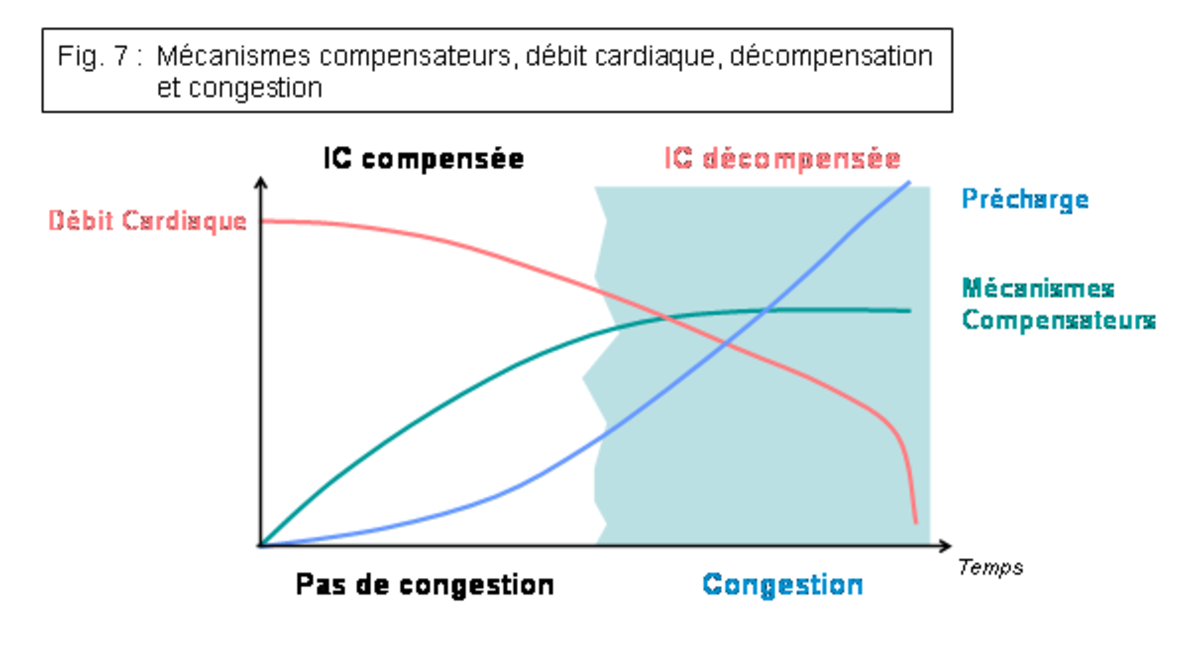
Tant que les mécanismes compensateurs permettent de maintenir un débit cardiaque et une perfusion tissulaire suffisants, il n’y a aucun symptôme clinique ; l’insuffisance cardiaque est dite asymptomatique ou compensée. Lorsque les mécanismes compensateurs sont dépassés, la perfusion tissulaire et le débit cardiaque ne peuvent plus être assurés et les premiers symptômes cliniques commencent à apparaître. C’est la phase d’Insuffisance Cardiaque Décompensée ou Symptomatique. Dans la très grande majorité des cas, les symptômes sont d’abord liés à l’augmentation de la précharge, qui se traduit par une stase veineuse en amont du cœur, avec dilatations vasculaires, œdème pulmonaire et épanchements pleural et/ou abdominal. On parle de congestion ; l’insuffisance cardiaque est alors dite Congestive. Dans la majorité des cas, l’insuffisance cardiaque est d’abord gauche. La congestion se manifeste donc primitivement dans le territoire pulmonaire.
Les classifications de l’insuffisance cardiaque
Afin de faciliter l’évaluation et le suivi de l’insuffisance cardiaque, divers classifications ont été proposées. Actuellement, les deux les plus utilisées sont la NYHA (New-York Heart Association) modifiée et l’ISACHC (International Small Animal Cardiac Health Council). Ces deux classifications font la différence entre une classe 1 asymptomatique, et les stades suivants représentant les insuffisances cardiaques congestives.


















































