Dystrophie myotonique de Steinert - Définition
La liste des auteurs de cet article est disponible ici.
Introduction
| ||||
|---|---|---|---|---|
| Autre nom | Maladie de Steinert | |||
| Référence MIM | 160900 | |||
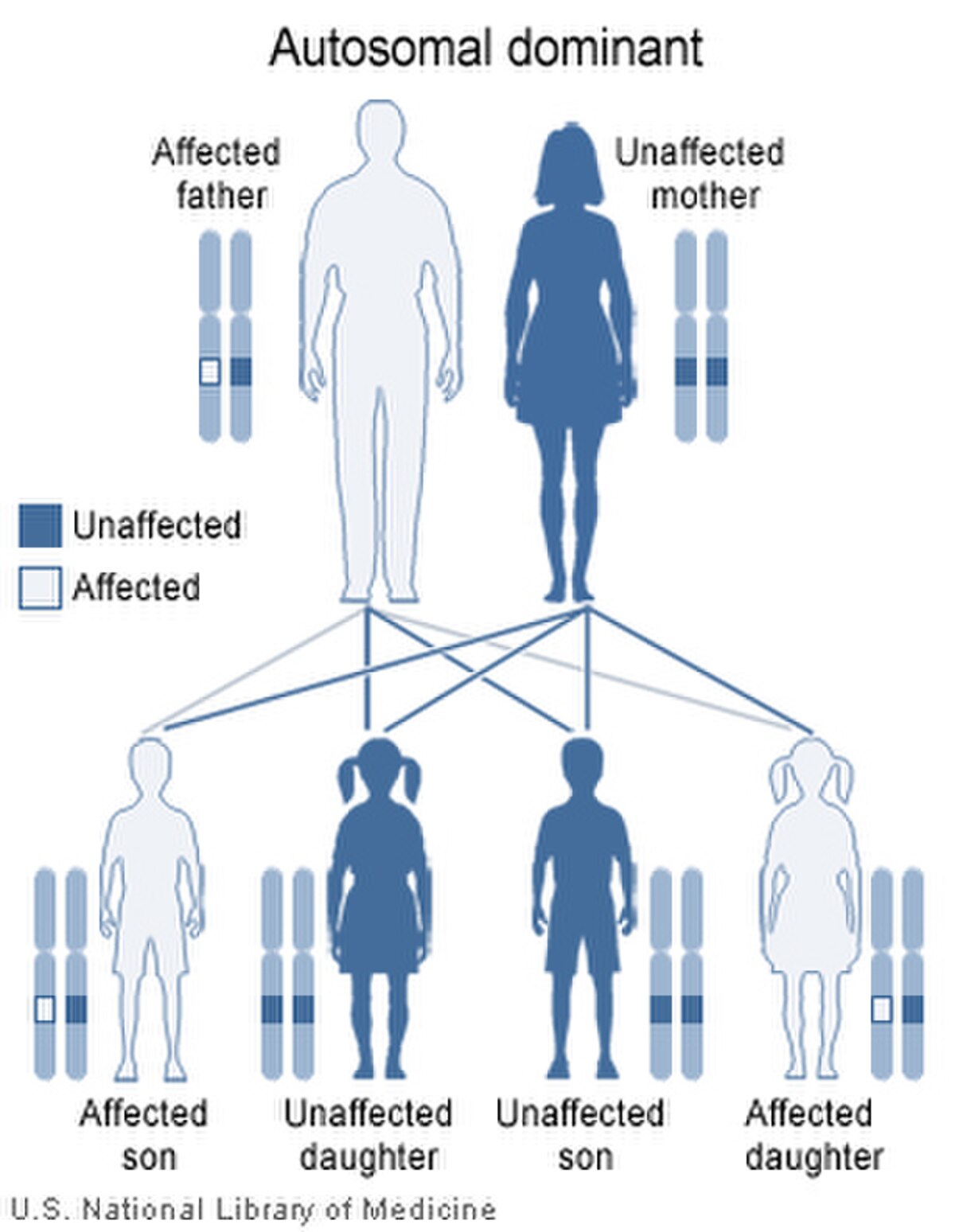
| Transmission | Dominante | |||
| Chromosome | 19 q13.2-q13.3 | |||
| Gène | DMPK | |||
| Empreinte parentale | Non | |||
| Mutation | Expansion de triplet | |||
| Mutation de novo | Rare | |||
| Nombre d'allèles pathologiques | Sans objet | |||
| Anticipation | Oui maternelle | |||
| Porteur sain | Sans objet | |||
| Incidence | Inconnue | |||
| Prévalence | 1/20000 | |||
| Pénétrance | Inconnue | |||
| Nombre de cas | Inconnu | |||
| Maladie génétiquement liée | Aucune | |||
| Diagnostic prénatal | Possibe | |||
| Article principal | {{{Article}}} | |||
| Liste des maladies génétiques à gène identifié | ||||
La dystrophie myotonique de Steinert ou maladie de Steinert est une maladie génétique autosomique dominante, à pénétrance incomplète et marquée par l'anticipation, qui affecte plusieurs organes : le squelette, les muscles lisses, l'œil, le cœur, le système endocrinien et le système nerveux central.
Les signes de cette maladie sont variés allant d'une forme légère à grave. Trois formes sont habituellement décrites selon l'âge d'apparition des premiers symptômes mais dont les limites ne sont pas toujours nettes : légère, classique et congénitale.
- La forme légère est caractérisée par une cataracte et une myotonie modérée. L'espérance de vie est normale.
- La forme classique est caractérisée par une faiblesse musculaire généralisée et une myotonie généralisée, une cataracte et des troubles de la conduction cardiaque. L'adulte peut perdre son autonomie et l'espérance de vie est réduite si le patient n'est pas suivi pour le coeur.
- La forme congénitale avec une hypotonie musculaire, souvent associée à une insuffisance respiratoire avec décès précoce. Le retard mental est fréquent dans cette forme.
La mutation est une expansion instable d'un triplet CTG du gène DMPK. Ce triplet est répété plus de 37 fois chez les personnes atteintes. Le nombre de répétitions du triplet CTG est généralement associé à la sévérité de la maladie ainsi qu'à l'âge d'apparition des symptômes.
Autres noms
- Myotonie dystrophique de type 1
- Dystrophie myotonique de type 1
Prévalence & incidence
La prévalence de cette maladie est de 1 sur 100 000 au Japon et 1 sur 10 000 en Islande. La prévalence mondiale de cette maladie est de 1 sur 20 000. La prévalence mondiale la plus élevée atteint 189 sur 100 000 de population dans la région du Saguenay-Lac-Saint-Jean au Québec (Canada).
Étiologie
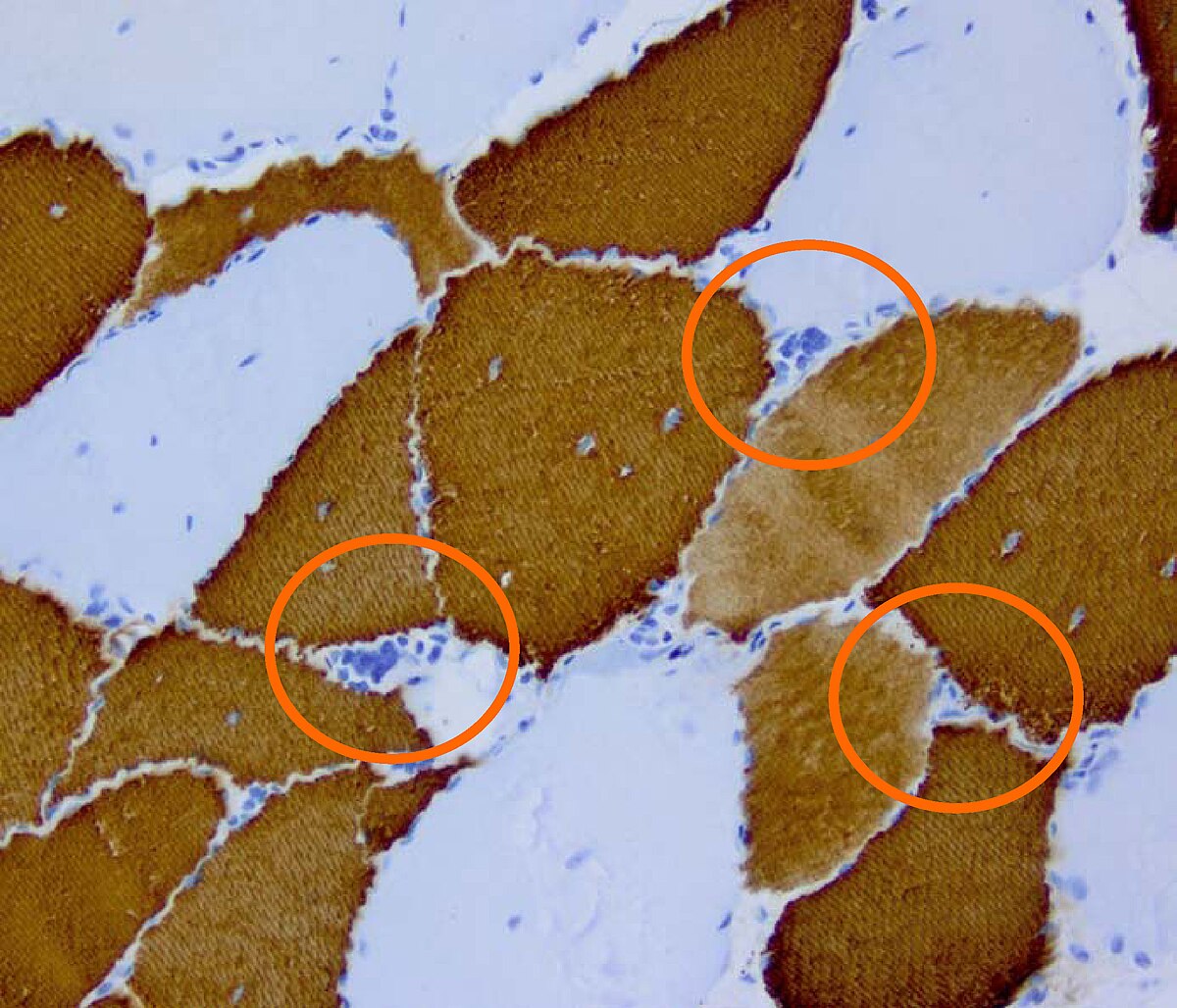
Mutation du gène DMPK (pour Dystrophy Myotonic Protein Kinase) situé sur le locus q13-2 chromosome 19 codant la myotonine, une Protéine Kinase cAMP dépendante, dont le rôle précis est inconnu. La mutation en cause est une expansion du triplet CTG dont le nombre dépasse 37 chez les personnes atteintes. Lorsque le nombre de répétition est supérieur à 50, la maladie se manifeste toujours. Dans la forme congénitale, l'expansion du triplet atteint plusieurs milliers.
Diagnostic
- La présomption du diagnostic repose sur l'association de signes cliniques variés (p. ex., cataracte, calvitie, troubles musculaires et cardiaques). La myotonie est le relachement lent de la contraction musculaire volontaire (main qui reste serrée après une poignée de main, par exemple).
- La certitude du diagnostic est obtenue par une technique de biologie moléculaire, avec un prélèvement sanguin. Le consentement de la personne prélevée est indispensable. La technique de TP-PCR est utilisée et suffit à éliminer les sujets sains (nombre de CTG inférieur à 37), hétérozygotes pour le nombre de répétitions du triplet CTG sur le gène DMPK (les deux allèles comportent moins de 37 répétitions). Chez les homozygotes sains et les hétérozygotes avec expansion (ils ont un allèle à moins de 37 répétitions, et un allèle à plus de 37, et généralement plus de 50 CTG), on réalise une vérification par Southern blot. Les hétérozygotes avec expansion sont atteints de la Dystrophie myotonique de Steinert.
- La présence d'un cas de Dystrophie de Steinert au sein d'une famille doit déclencher une enquête familiale. Là encore, l'enquête génétique n'est possible qu'avec l'accord explicite des personnes prélevées.
Différentiel
- Myotonie dystrophique type 2 secondaire à une expansion de CCTG du gène ZNF9
- Myopathie à corps d'inclusion
- Myopathie avec surcharge en desmine

















































