Apoptose - Définition
La liste des auteurs de cet article est disponible ici.
Manifestation cytologique
L'apoptose se manifeste sur ces cellules isolées (non regroupées). On constaste sur les cellules concernées une compaction et une marginalisation de la chromatine nucléaire ainsi qu'une convolution des membranes cytoplasmiques et nucléaires et une condensation du cytoplasme.
L'intégrité des membranes est conservée au cours du processus apoptotique évitant ainsi toute réaction inflammatoire. Les corps apoptotiques sont protégés par une enveloppe membranaire issue de la convolution des membranes. .
Mécanisme intracellulaire
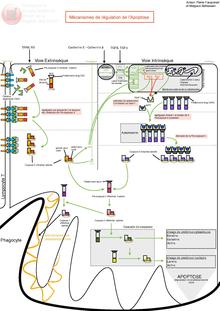
Le mécanisme d’apoptose est gouverné par deux voies principales d’activation :
- une voie dite extrinsèque, impliquant des récepteurs appartenant à la superfamille des récepteurs au TNF,
- une voie dite intrinsèque mettant particulièrement en jeu la mitochondrie ; cette voie est gouvernée par des protéines appartenant à la superfamille de Bcl-2.
Ces deux voies conduisent à l’activation de protéases à cystéine appelées caspases, responsables des phénomènes morphologiques et biochimiques observés : exposition de phosphatidylsérine à la surface de la membrane cellulaire, arrêt de la réplication, fragmentation du noyau et du cytosquelette entraînant la formation de corps apoptotiques phagocytés par les cellules environnantes.
Voie extrinsèque
Dans l'état des connaissances actuelles, il semble que le mécanisme d'activation du récepteur Fas puisse être étendu aux autres récepteurs de mort. La signalisation par Fas recrute un complexe composé d'une molécule adaptatrice FADD (Fas Associated Death Domain) et de la procaspase-8. FADD se lie à travers son propre domaine de mort à ceux des récepteurs Fas. FADD contient également un domaine qui se lie à la procaspase-8. La formation de ce complexe entraîne le clivage de la caspase-8 qui est alors produite sous sa forme dimérique active, puis la cascade d'activation séquentielle des différentes caspases parmi lesquelles la caspase-3. Ces différentes protéases effectuent le clivage de plusieurs molécules, parmi lesquelles des protéines de structure et des protéines impliquées dans les systèmes de réparation cellulaire. Il existe 2 voies de transduction du signal de la voie Fas, dépendant du type cellulaire. Dans les cellules de type 1 comme les thymocytes, la caspase 8 active directement la caspase 3. Dans les cellules de type 2 comme les hépatocytes, la caspase 8 active Bid, provoquant la libération du cytochrome c. L'association cytochrome c/APAF1 active la caspase 9 qui active à son tour la caspase 3.
Voie intrinsèque
La grande famille des protéines homologues à Bcl-2 joue un rôle majeur dans la régulation de l'apoptose. Cette régulation passe par la modulation de l'activité de certaines caspases, principalement la caspase 9. Ainsi, en empêchant la libération du cytochrome c par la mitochondrie, Bcl-2 et Bcl-XL inhibent la formation du complexe APAF1/cytochrome c/procaspase 9 nécessaire à l'apoptose. La mitochondrie joue un rôle clé dans la régulation de l'apoptose. En effet, la phase effectrice de l'apoptose comporte l'ouverture des pores de transition de perméabilité des mitochondries. Ces pores sont des canaux oligo-protéiques constitués au niveau de la membrane externe par la porine (ou VDAC : Voltage Dependent Anion Channel), sur la membrane interne par l'ANT (Adenine Nucléotide Translocator). Suite à l’ouverture de ces pores, il y a libération de molécules pro-apoptotiques telles que le cytochrome c, les caspases 2, 3 et 9 ainsi que l’Apoptosis Inducing Factor (AIF). L’AIF est une des molécules pro-apoptotiques libérées de la mitochondrie, il est localisé dans l'espace intermembranaire mitochondrial. Il s'agit d'une molécule possédant une double fonction : oxydoréductase et facteur pro-apoptotique. Afin que cette dernière activité s'exerce, il y a nécessité d'une redistribution subcellulaire : de la mitochondrie vers le cytosol puis vers le noyau cellulaire. La voie AIF est indépendante des caspases et ne nécessite aucun intermédiaire pour provoquer l'apoptose nucléaire : il interviendrait dans la voie apoptotique indépendante des caspases. Le mécanisme central de cette phase effectrice est l'altération de la perméabilité membranaire mitochondriale et l'ouverture du PTPC (Permeability Transition Pore Complex), un complexe multiprotéique de la membrane interne mitochondriale. Cette phase s'accompagne d'une diminution du potentiel transmembranaire mitochondrial (Delta Psi m), suivi du gonflement de la matrice mitochondriale, une interruption du métabolisme énergétique aérobique et un stress oxydatif. Par voie de conséquence, des protéines intermembranaires telles que la protéine AIF, le cytochrome c, certaines pro-caspases, l'endonucléase G et d'autres facteurs vont être relargués dans le cytosol, initiant la phase de dégradation. Cette phase de libération est sous le contrôle de membres de la famille Bcl-2. Ainsi, Bcl-2 est capable de bloquer la sortie du cytochrome c alors que Bax peut l'induire. Dans la majorité des cas la libération du cytochrome c est indépendante de l'activité des caspases. L'activation des caspases induite par le cytochrome c cytosolique associé à APAF1 ou l'apoptose induite par les récepteurs de mort ne sont pas des mécanismes totalement indépendants. En effet, Bid est le lien entre les récepteurs de mort et la libération du cytochrome c. Bid est directement clivé par la caspase 8 et le fragment C-terminal produit permet la libération du cytochrome c. Ainsi, des extraits cytosoliques déplétés en Bid rendent la caspase 8 incapable de libérer le cytochrome c in vitro. En parallèle, des études ont montré que l'addition de molécules recombinantes Bax ou Bak sur des mitochondries isolées pouvait induire la libération du cytochrome c ainsi que la perte du potentiel transmembranaire mitochondrial.

















































