Restez toujours informé: suivez-nous sur Google Actualités (icone ☆)
Comment sélectionner des médicaments avant même les essais cliniques ? Des chimistes de l'Institut de chimie organique et analytique (ICOA, CNRS/Université d'Orléans) et du Centre de biophysique moléculaire (CBM, CNRS) proposent un nouveau modèle in silico, qui décrit la durée des interactions entre une molécule et sa cible biologique. Publiés dans le Journal of Chemical Information and Modeling, ces travaux ont prédit avec succès des effets sur une protéine liée à certains cancers et aident à diminuer les doses et ainsi la toxicité.
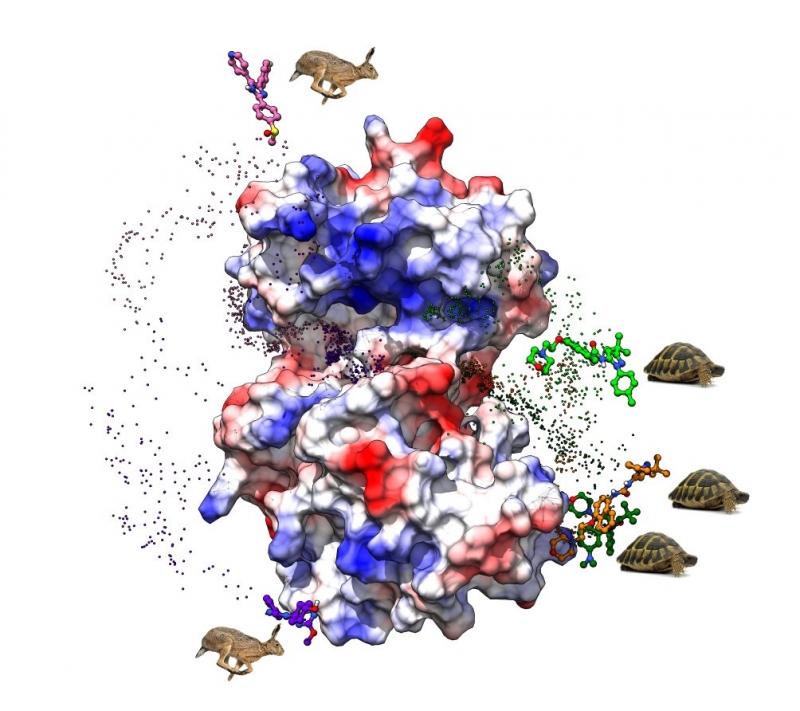
Au centre, la protéine kinase p38a. Les dynamiques de différentes molécules sont testées, certains sont rapides (les lièvres), d'autres lentes (les tortues). © Aci-Sèche
Avant leur mise sur le marché, les médicaments passent par de nombreuses phases de test. Leur évaluation en amont reste difficile, si bien que nombre d'entre eux échouent lors des essais cliniques. Afin de mieux sélectionner les molécules thérapeutiques, des chercheurs de l'Institut de chimie organique et analytique (ICOA, CNRS/Université d'Orléans) et du Centre de biophysique moléculaire (CBM, CNRS) ont développé une méthode in silico qui simule le temps de résidence entre le médicament et sa cible biologique. Cela a pour objectif d'améliorer l'efficacité du médicament lors des essais cliniques de phase II, et de diminuer ainsi le taux d'échec.
Les précédentes études se concentraient sur les relations structure-activité, c'est-à-dire comment la structure chimique de la molécule affecte son interaction avec sa cible. Mais de nouveaux travaux, comme ceux-ci, s'attachent davantage aux relations structure-cinétique (SKR) qui décrivent le temps de résidence, soit le temps pendant lequel la molécule reste en interaction avec sa cible. Le modèle proposé par les chercheurs utilise des simulations de dynamique moléculaire biaisée, où s'appliquent des contraintes artificielles et contrôlées. Cela permet d'accélérer la simulation du processus biologique réel, ces processus pouvant s'étaler sur plusieurs jours. L'équipe a ensuite simulé divers cas concernant la protéine kinase p38a, impliquée dans certains cancers et maladies neurodégénératives. Elle a de plus été choisie car c'est une des rares protéines kinases pour laquelle des résultats expérimentaux de SKR étaient déjà disponibles. Le modèle a alors fourni des valeurs théoriques cohérentes par rapport à ces données expérimentales. Les chercheurs comptent tester leur simulation sur d'autres molécules et protéines.
Référence:
Braka A., Garnier N., Bonnet P., Aci-Sèche S.Residence Time Prediction of Type 1 and 2 Kinase Inhibitors from Unbinding Simulations. J Chem Inf Model. 6 janvier 2020.
doi: 10.1021/acs.jcim.9b00497
Contact:
- Samia Aci-Sèche - Institut de chimie organique et analytique (ICOA) - samia.aci-seche at univ-orleans.fr
- Pascal Bonnet - Institut de chimie organique et analytique (ICOA) - pascal.bonnet at univ-orleans.fr
- Stéphanie Younès - Responsable Communication - inc.communication at cnrs.fr
Populaires